DNM2 Complex Regulates Leukemic Properties of Primitive CML Cells Through Enhanced Cellular Endocytosis and ROS-Mediated Autophagy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dynamin Functions and Ligands: Classical Mechanisms Behind
1521-0111/91/2/123–134$25.00 http://dx.doi.org/10.1124/mol.116.105064 MOLECULAR PHARMACOLOGY Mol Pharmacol 91:123–134, February 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics MINIREVIEW Dynamin Functions and Ligands: Classical Mechanisms Behind Mahaveer Singh, Hemant R. Jadhav, and Tanya Bhatt Department of Pharmacy, Birla Institute of Technology and Sciences Pilani, Pilani Campus, Rajasthan, India Received May 5, 2016; accepted November 17, 2016 Downloaded from ABSTRACT Dynamin is a GTPase that plays a vital role in clathrin-dependent pathophysiology of various disorders, such as Alzheimer’s disease, endocytosis and other vesicular trafficking processes by acting Parkinson’s disease, Huntington’s disease, Charcot-Marie-Tooth as a pair of molecular scissors for newly formed vesicles originating disease, heart failure, schizophrenia, epilepsy, cancer, dominant ’ from the plasma membrane. Dynamins and related proteins are optic atrophy, osteoporosis, and Down s syndrome. This review is molpharm.aspetjournals.org important components for the cleavage of clathrin-coated vesicles, an attempt to illustrate the dynamin-related mechanisms involved phagosomes, and mitochondria. These proteins help in organelle in the above-mentioned disorders and to help medicinal chemists division, viral resistance, and mitochondrial fusion/fission. Dys- to design novel dynamin ligands, which could be useful in the function and mutations in dynamin have been implicated in the treatment of dynamin-related disorders. Introduction GTP hydrolysis–dependent conformational change of GTPase dynamin assists in membrane fission, leading to the generation Dynamins were originally discovered in the brain and identi- of endocytic vesicles (Praefcke and McMahon, 2004; Ferguson at ASPET Journals on September 23, 2021 fied as microtubule binding partners. -

Targeted Genes and Methodology Details for Neuromuscular Genetic Panels
Targeted Genes and Methodology Details for Neuromuscular Genetic Panels Reference transcripts based on build GRCh37 (hg19) interrogated by Neuromuscular Genetic Panels Next-generation sequencing (NGS) and/or Sanger sequencing is performed Motor Neuron Disease Panel to test for the presence of a mutation in these genes. Gene GenBank Accession Number Regions of homology, high GC-rich content, and repetitive sequences may ALS2 NM_020919 not provide accurate sequence. Therefore, all reported alterations detected ANG NM_001145 by NGS are confirmed by an independent reference method based on laboratory developed criteria. However, this does not rule out the possibility CHMP2B NM_014043 of a false-negative result in these regions. ERBB4 NM_005235 Sanger sequencing is used to confirm alterations detected by NGS when FIG4 NM_014845 appropriate.(Unpublished Mayo method) FUS NM_004960 HNRNPA1 NM_031157 OPTN NM_021980 PFN1 NM_005022 SETX NM_015046 SIGMAR1 NM_005866 SOD1 NM_000454 SQSTM1 NM_003900 TARDBP NM_007375 UBQLN2 NM_013444 VAPB NM_004738 VCP NM_007126 ©2018 Mayo Foundation for Medical Education and Research Page 1 of 14 MC4091-83rev1018 Muscular Dystrophy Panel Muscular Dystrophy Panel Gene GenBank Accession Number Gene GenBank Accession Number ACTA1 NM_001100 LMNA NM_170707 ANO5 NM_213599 LPIN1 NM_145693 B3GALNT2 NM_152490 MATR3 NM_199189 B4GAT1 NM_006876 MYH2 NM_017534 BAG3 NM_004281 MYH7 NM_000257 BIN1 NM_139343 MYOT NM_006790 BVES NM_007073 NEB NM_004543 CAPN3 NM_000070 PLEC NM_000445 CAV3 NM_033337 POMGNT1 NM_017739 CAVIN1 NM_012232 POMGNT2 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Conserved and Novel Properties of Clathrin-Mediated Endocytosis in Dictyostelium Discoideum" (2012)
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2012 Conserved and Novel Properties of Clathrin- Mediated Endocytosis in Dictyostelium Discoideum Laura Macro Follow this and additional works at: http://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons Recommended Citation Macro, Laura, "Conserved and Novel Properties of Clathrin-Mediated Endocytosis in Dictyostelium Discoideum" (2012). Student Theses and Dissertations. Paper 163. This Thesis is brought to you for free and open access by Digital Commons @ RU. It has been accepted for inclusion in Student Theses and Dissertations by an authorized administrator of Digital Commons @ RU. For more information, please contact [email protected]. CONSERVED AND NOVEL PROPERTIES OF CLATHRIN- MEDIATED ENDOCYTOSIS IN DICTYOSTELIUM DISCOIDEUM A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Laura Macro June 2012 © Copyright by Laura Macro 2012 CONSERVED AND NOVEL PROPERTIES OF CLATHRIN- MEDIATED ENDOCYTOSIS IN DICTYOSTELIUM DISCOIDEUM Laura Macro, Ph.D. The Rockefeller University 2012 The protein clathrin mediates one of the major pathways of endocytosis from the extracellular milieu and plasma membrane. Clathrin functions with a network of interacting accessory proteins, one of which is the adaptor complex AP-2, to co-ordinate vesicle formation. Disruption of genes involved in clathrin-mediated endocytosis causes embryonic lethality in multicellular animals suggesting that clathrin-mediated endocytosis is a fundamental cellular process. However, loss of clathrin-mediated endocytosis genes in single cell eukaryotes, such as S.cerevisiae (yeast), does not cause lethality, suggesting that clathrin may convey specific advantages for multicellularity. -

Myopathy Genes (HGNC) Neuropathy (HGNC) Neuromuscular Disease
Myopathy Genes Neuropathy Neuromuscular Disease (HGNC) (HGNC) (HGNC) ABHD5 ABCA1 ADCK3 ACTG2 ACO2 AGRN AGK AGXT ALS2 ALDOA AIFM1 ANG AMER1 ALAD AP4B1 ANO5 AMACR AP4E1 AR AP1S1 AP4M1 AUH APTX AP4S1 B4GALT1 AR AP5Z1 CACNA1S ATL3 ATM CASQ1 B4GALNT1 ATXN10 CCDC78 BAG3 ATXN7 CHCHD10 BRP44L BEAN1 CHRNA1 C12orf65 C9orf72 CHRNB1 C19orf12 CACNB4 CHRND C1NH CAPN3 CHRNE CECR1 CHAT CLPB CISD2 CHKB COL6A1 CLCF1 CHMP2B COL6A2 CLCN2 CHRNG COL6A3 CLP1 CLCN1 COLQ CMT2G COL9A3 CTNS CMT2H COQ2 DGUOK CMTDIA COQ6 DNA2 CMTX2 COQ9 DNAJB6 CMTX3 COX15 DNAJC19 COASY CPT1A DNM2 COX6A1 CYP7B1 DPM2 CPOX DAG1 DYSF CYP27A1 DDHD2 EMD CYP2U1 DOK7 EPG5 DARS2 DPAGT1 FAM111B DCAF8 DPM3 FBXL4 DDHD1 DUX4 FKBP14 DFNX5 ECEL1 FKRP DHTKD1 ERBB3 FLH1 DIAPH3 ERLIN2 FLNC DNAJB2 FA2H HNRNPA1 DNAJC3 FKTN HNRNPDL ELOVL5 FUS HNRPA2B1 ERCC8 G6PC KLHL40 FAH GFPT1 KLHL41 FAM126A GLE1 LAMA2 FBN1 GYS2 LDB3 FMR1 HSPD1 LMOD3 FXN IFRD1 MEGF10 GALC INF2 MGME1 GBE1 ISPD MTAP GJC2 ITGA7 MTMR14 GP1BA ITPR1 MYF6 HADHA KCNA1 MYH14 HADHB KCNC3 MYLK2 HFE KCNE3 NARS2 HINT1 KCNJ18 NEB HK1 KCNJ2 ORAI1 HMBS KIAA0196 PRKAG2 HSD17B4 KIF21A PTEN HSN1B L1CAM RBCK1 IARS2 LAMB2 RET IGHMBP2 LARGE RMND1 KCNJ10 MCCC2 SCN4A KIF5A MRE11A SERAC1 LRSAM1 MRPL3 SGCA LYST MTO1 SIL1 MANBA MTPAP SPEG MARS MTTP STAC3 MTATP6 MUSK STIM1 MYH14 MYBPC3 SYNE1 MYOT MYH3 SYNE2 NAMSD MYH8 TAZ NF2 NF1 TIA1 NGLY1 NIPA1 TMEM43 NMSR NOP56 TNPO3 NOTCH3 OPTN TNXB OPA1 PDSS2 TPM2 OPA3 PDYN TRPV4 OTOF PFN1 UBA1 PDK3 PHKA2 VCP PDSS1 PHKG2 XDH PEX10 PHOX2A ACADS PEX2 PIP5K1C ACADVL PMM2 PLEC ACTA1 PNPLA6 PLP1 AGL PPOX POMGNT1 AMPD1 PRICKLE1 -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

A Centronuclear Myopathy-Dynamin 2 Mutation Impairs Skeletal Muscle Structure and Function in Mice
Human Molecular Genetics, 2010, Vol. 19, No. 24 4820–4836 doi:10.1093/hmg/ddq413 Advance Access published on September 21, 2010 A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice Anne-Ce´cile Durieux1,2,3,4, Alban Vignaud1,2,3,4, Bernard Prudhon1,2,3,4, Mai Thao Viou1,2,3,4, Maud Beuvin1,2,3,4, Ste´phane Vassilopoulos1,2,3,4, Bodvae¨l Fraysse1,2,3,4, Arnaud Ferry1,2,3,4, ∗ Jeanne Laine´ 1,2,3,4, Norma B. Romero 1,2,3,4,5, Pascale Guicheney1,6 and Marc Bitoun1,2,3,4, Downloaded from https://academic.oup.com/hmg/article/19/24/4820/680275 by guest on 25 September 2021 1Universite´ Pierre et Marie Curie-Paris 6, IFR14, Paris F-75013, France, 2CNRS, UMR 7215, Paris, France, 3Inserm, U974, Paris F-75013, France, 4Institut de Myologie, Paris, France, 5AP-HP, Groupe Hospitalier Pitie´-Salpeˆtrie`re, Paris F-75013, France and 6Inserm, U956, Paris F-75013, France Received July 1, 2010; Revised and Accepted September 16, 2010 Autosomal dominant centronuclear myopathy (AD-CNM) is due to mutations in the gene encoding dynamin 2 (DNM2) involved in endocytosis and intracellular membrane trafficking. To understand the pathomechanisms resulting from a DNM2 mutation, we generated a knock-in mouse model expressing the most frequent AD- CNM mutation (KI-Dnm2R465W). Heterozygous (HTZ) mice developed a myopathy showing a specific spatial and temporal muscle involvement. In the primarily and prominently affected tibialis anterior muscle, impair- ment of the contractile properties was evidenced at weaning and was progressively associated with atrophy and histopathological abnormalities mainly affecting mitochondria and reticular network. -

Huntington's Disease Associated Genes
Open Access Austin Journal of Genetics and Genomic Research Special Article - Genetics of Huntington’s Disease Huntington’s Disease Associated Genes: Molecular Basis of Disease and Possible New Targets for Treatment Bhattacharyya NP* BioMedical Genomics Centre, India Abstract *Corresponding author: Nitai P Bhattacharyya, Diverse molecular defects in Huntington’s Disease (HD) have been identified BioMedical Genomics Centre, PG Polyclinics (3rd Floor), 5 since the discovery of Huntingtin (HTT) gene mutated in the diseasein1993. Suburban Hospital Road, Kolkata 700 20, India Presently, there is no cure for the disease. To identify genes involved in HD pathogenesis, we collate data from diverse sources and using stringent criteria Received: June 22, 2016; Accepted: November 11, collected 523 HD associated genes. Enrichment analysis with these proteins 2016; Published: November 18, 2016 revealed that diverse biological processes and pathways like gene expression, apoptosis, proteasomal degradation, glucose/carbohydrate metabolism, known to involve in HD pathogenesis, were significantly enriched. HD associated genes were significantly over represented in biological processes like cell cycle and differentiation indicating their possible role in HD pathogenesis. Comparisons of HD associated genes with targets of FDA approved drugs and probable protein targets having similar properties as that of FDA approved protein targets of drugs, we identified 49 HD associated genes that could be new probable drug targets. HD associated genes collected here will be useful to decipher molecular basis of HD including regulation of these genes and new targets for the treatment of presently incurable devastating HD. Keywords: Huntington’s disease; Huntingtin interacting proteins; Co- expressed genes; Drug targets Abbreviations skeletal muscle loss. -

Supplementary Table S2
Table 2 HIF-1 target genes Unigene ID Hyp/Norm Name Name Hs.296323 0.50 SGK serum/glucocorticoid regulated kinase Hs.1048 0.53 KITLG KIT ligand Hs.1048 0.53 KITLG KIT ligand Hs.171723 0.55 TAF9L TAF9-like RNA polymerase II, TATA box binding protein Hs.374524 0.55 DHX9 DEAH (Asp-Glu-Ala-His) box polypeptide 9 Hs.21486 0.56 STAT1 signal transducer and activator of transcription 1, 91kDa Hs.390504 0.57 SOS2 son of sevenless homolog 2 (Drosophila) Hs.2227 0.57 CEBPG CCAAT/enhancer binding protein (C/EBP), gamma Hs.464526 0.58 ABHD3 abhydrolase domain containing 3 Hs.107056 0.60 CED-6 PTB domain adaptor protein CED-6 Hs.417157 0.61 MGC14376 hypothetical protein MGC14376 Hs.201939 0.61 ATP6V0A2 ATPase, H+ transporting, lysosomal V0 subunit a isoform 2 Hs.7879 0.61 IFRD1 interferon-related developmental regulator 1 Hs.25204 0.61 CHST12 carbohydrate (chondroitin 4) sulfotransferase 12 Hs.58617 0.62 ROCK2 Rho-associated, coiled-coil containing protein kinase 2 Hs.477132 0.62 COPA coatomer protein complex, subunit alpha Hs.389893 0.62 DLG1 discs, large homolog 1 (Drosophila) Hs.439052 0.63 SPIN spindlin Hs.334534 0.63 GNS glucosamine (N-acetyl)-6-sulfatase (Sanfilippo disease IIID) Hs.459631 0.63 Homo sapiens cDNA FLJ12835 fis, clone NT2RP2003165. Hs.336425 0.63 Homo sapiens mRNA; cDNA DKFZp313P052 Hs.17969 0.64 KIAA0663 KIAA0663 gene product Hs.28578 0.65 MBNL1 muscleblind-like (Drosophila) Hs.55220 0.65 BAG2 BCL2-associated athanogene 2 Hs.143840 0.65 KIAA0852 KIAA0852 protein Hs.432996 0.66 FLJ12448 hypothetical protein FLJ12448 Hs.159118 0.66 -

A Centronuclear Myopathy
A Centronuclear Myopathy - Dynamin 2 Mutation Impairs Autophagy in Mice Anne-Cécile Durieux, Stéphane Vassilopoulos, Jeanne Lainé, Bodvael Fraysse, Laura Briñas, Bernard Prudhon, Josiane Castells, Damien Freyssenet, Gisele Bonne, Pascale Guicheney, et al. To cite this version: Anne-Cécile Durieux, Stéphane Vassilopoulos, Jeanne Lainé, Bodvael Fraysse, Laura Briñas, et al.. A Centronuclear Myopathy - Dynamin 2 Mutation Impairs Autophagy in Mice. Traffic, Wiley, 2012, 13 (6), pp.869-879. 10.1111/j.1600-0854.2012.01348.x. hal-02453822 HAL Id: hal-02453822 https://hal.archives-ouvertes.fr/hal-02453822 Submitted on 24 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. A centronuclear myopathy-dynamin 2 mutation impairs autophagy in mice Anne-Cécile Durieux1,2,3,4, Stéphane Vassilopoulos1,2,3,4, Jeanne Lainé1,2,3,4,5, Bodvaël Fraysse1,2,3,4, Laura Briñas1,2,3,4, Bernard Prudhon1,2,3,4, Josiane Castells6, Damien Freyssenet6, Gisèle Bonne1,2,3,4,7, Pascale Guicheney8, Marc Bitoun1,2,3,4. 1 Université Pierre et Marie Curie-Paris 6, UM76, Paris, F-75013, France. 2 Inserm, U974, Paris, F-75013, France. 3 CNRS, UMR 7215, Paris, France. -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Autism and Intellectual Disability TRIO Panel Test Code TR002 Test Summary This test analyzes 2429 genes that have been associated with Autism and Intellectual Disability and/or disorders associated with Autism and Intellectual Disability with the analysis being performed as a TRIO Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing Comprehensive test for patients with intellectual disability or global developmental delays (Moeschler et al 2014 PMID: 25157020). Comprehensive test for individuals with multiple congenital anomalies (Miller et al. 2010 PMID 20466091). Patients with autism/autism spectrum disorders (ASDs). Suspected autosomal recessive condition due to close familial relations Previously negative karyotyping and/or chromosomal microarray results. Test Description This panel analyzes 2429 genes that have been associated with Autism and ID and/or disorders associated with Autism and ID. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Autism Spectrum Disorder (ASD) refers to a group of developmental disabilities that are typically associated with challenges of varying severity in the areas of social interaction, communication, and repetitive/restricted behaviors. -
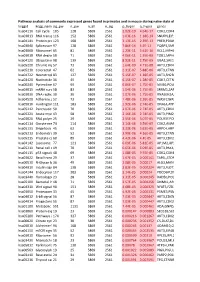
Pathway Analysis of Commonly Expressed Genes Found in Primates and in Mouse During Naïve State of Pluripotenc Keggid Kegg Names
Pathway analysis of commonly expressed genes found in primates and in mouse during naïve state of pluripotency. keggid kegg_namesig_pw n_pw n_all n_sig p_hyper q_hyper genes hsa04110 Cell cycle 105 128 5869 2561 2.02E-19 4.64E-17 CDK2,CDK4,CDK6,CDK7,CDKN1A,CDKN1B,STAG1,CDKN2B,ANAPC10,MAD2L2,STAG2,GADD45G,DBF4,YWHAQ,CHEK1,CHEK2,CREBBP,GADD45A,E2F1,E2F3,E2F4,E2F5,EP300,ORC3,CDC26,ABL1,ANAPC13,SFN,GSK3B,ANAPC2,ANAPC4,HDAC1,HDAC2,MAD2L1,SMAD2,SMAD3,SMAD4,MCM2,MCM3,MCM4,MCM5,MCM6,MCM7,MDM2,MYC,GADD45B,ATM,ORC1,ORC2,ORC4,ORC5,FZR1,ANAPC7,ANAPC11,PLK1,ATR,PRKDC,RAD21,RB1,RBL1,CCND1,ANAPC1,SKP2,BUB1,BUB1B,TFDP1,TFDP2,TGFB1,TGFB2,TTK,WEE1,YWHAB,YWHAE,YWHAG,YWHAH,YWHAZ,ZBTB17,SMC1A,CDC7,CDC45,MAD1L1,CDC14A,CDC23,CDC16,CCNA2,CCNB1,CCND2,CCND3,CCNE1,CCNH,PKMYT1,SMC3,CCNB2,CCNE2,BUB3,PTTG1,ESPL1,CDK1,CDC6,CDC20,CDC25A,CDC25B,CDC25C,CDC27,RBX1 hsa03013 RNA transport116 152 5869 2561 1.03E-16 1.18E-14 SNUPN,EIF1,POP7,SRRM1,SAP18,EIF1B,PRMT5,TACC3,NXF1,RPP30,RPP38,PAIP1,POP4,RPP40,RNPS1,POP1,STRAP,DDX20,XPOT,CLNS1A,NUP35,RPP25L,EEF1A1,EEF1A2,EIF2S1,EIF2B1,EIF4A1,EIF4B,EIF4E,EIF4EBP1,EIF4G1,EIF4G2,EIF5,CASC3,NCBP2,ACIN1,NUP205,NUP210,NUP62,GEMIN5,UPF2,PABPC1,NXT1,NUP43,EIF3E,KPNB1,MAGOH,NCBP1,NUP88,NUP98,GEMIN4,NMD3,POP5,PHAX,NUP54,PNN,RPP25,GEMIN8,MAGOHB,ELAC1,NDC1,NUP133,NXT2,NUP107,THOC2,XPO5,RAN,RANBP2,RANGAP1,SENP2,UPF1,ELAC2,SEC13,UPF3B,SMN1,SUMO3,SUMO2,TPR,SUMO1,XPO1,NUP37,DDX39B,THOC6,GEMIN7,GEMIN6,RPP21,NUP85,THOC7,NUP214,AAAS,SEH1L,THOC3,RAE1,THOC5,EIF3A,EIF3B,EIF3C,EIF3D,EIF3F,EIF3G,EIF3H,EIF3I,EIF4G3,PABPC4,EIF2B4,EIF2B2,EIF2B5,EIF2S2,EIF4E2,NUP155,EIF5B,TGS1,NUP93,EIF4A3,NUP153,THOC1