2011TOU30118.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Safety Data Sheet
SAFETY DATA SHEET SECTION 1: PRODUCT IDENTIFICATION PRODUCT NAME DHEA (Prasterone) (Micronized) PRODUCT CODE 0733 SUPPLIER MEDISCA Inc. Tel.: 1.800.932.1039 | Fax.: 1.855.850.5855 661 Route 3, Unit C, Plattsburgh, NY, 12901 3955 W. Mesa Vista Ave., Unit A-10, Las Vegas, NV, 89118 6641 N. Belt Line Road, Suite 130, Irving, TX, 75063 MEDISCA Pharmaceutique Inc. Tel.: 1.800.665.6334 | Fax.: 514.338.1693 4509 Rue Dobrin, St. Laurent, QC, H4R 2L8 21300 Gordon Way, Unit 153/158, Richmond, BC V6W 1M2 MEDISCA Australia PTY LTD Tel.: 1.300.786.392 | Fax.: 61.2.9700.9047 Unit 7, Heritage Business Park 5-9 Ricketty Street, Mascot, NSW 2020 EMERGENCY PHONE CHEMTREC Day or Night Within USA and Canada: 1-800-424-9300 NSW Poisons Information Centre: 131 126 USES Adjuvant; Androgen SECTION 2: HAZARDS IDENTIFICATION GHS CLASSIFICATION Toxic to Reproduction (Category 2) PICTOGRAM SIGNAL WORD Warning HAZARD STATEMENT(S) Reproductive effector, prohormone. Suspected of damaging fertility or the unborn child. May cause harm to breast-fed children. Causes serious eye irritation. Causes skin and respiratory irritation. Very toxic to aquatic life with long lasting effects. AUSTRALIA-ONLY HAZARDS Not Applicable. PRECAUTIONARY STATEMENT(S) Prevention Wash thoroughly after handling. Obtain special instructions before use. Do not handle until all safety precautions have been read and understood. Do not breathe dusts or mists. Do not eat, drink or smoke when using this product. Avoid contact during pregnancy/while nursing. Wear protective gloves, protective clothing, eye protection, face protection. Avoid release to the environment. Response IF ON SKIN (HAIR): Wash with plenty of water. -

Vargas KEA, Et Al. Hepatotoxicity Associated with Methylstenbolone and Copyright© Vargas KEA, Et Al
1. Medical Journal of Clinical Trials & Case Studies ISSN: 2578-4838 Hepatotoxicity Associated with Methylstenbolone and Stanozolol Abuse Vargas KEA*, Guaraná TA, Biccas BN, Agoglia LV, Carvalho ACG, Case Report Gismondi R and Esberard EBC Volume 2 Issue 5 Received Date: July 27, 2018 Department of Gastroenterology/Hepatology, Department of Clinical Medicine, and Published Date: September 03, 2018 Department of Pathology, Antônio Pedro University Hospital, Federal Fluminense DOI: 10.23880/mjccs-16000176 University, Rio de Janeiro, Brazil *Corresponding author: Vargas Karen Elizabeth Arce, Department of Gastroenterology/Hepatology, Department of Clinical Medicine, and Department of Pathology, Antônio Pedro University Hospital, Federal Fluminense University, Rio de Janeiro, Ernani do Amaral Peixoto Avenue, 935. Ap.901 / Cep.24020043, Brazil, Tel: 005521981584624; Email: [email protected] Abstract Background & Objectives: Drug hepatotoxicity is a major cause of liver disease. Many drugs are well known to induce liver damage. Some toxic products, like anabolic androgenic steroids, that are pharmaceutical preparations since they contain pharmaceutically active substance, are available as nutritional supplements. Many patients are used to consume these like dietary stuff. Methods: We introduce a case series of two patients who developed hepatic damage after the consumption of anabolic- androgenic steroids, accompanied by a detailed bibliographic research on this topic. Results: We present two young men who developed significant liver damage, both with hyperbilirubinemia pattern after consumption of anabolic-androgenic steroids. This was associated with considerable morbidity, although both recovered without liver transplantation. The two anabolic-androgenic steroids were being marketed as dietary supplements. Conclusions: Although not well controlled substances in Brazil, anabolic-androgenic steroids are cause of severe hepatotoxicity. -

Records of Pharmaceutical and Biomedical Sciences
REVIEW ARTICLE RECORDS OF PHARMACEUTICAL AND BIOMEDICAL SCIENCES Effect of Exogenous Anabolic Androgenic Steroids on Testosterone/ Epitestosterone Ratio and its Application on Athlete Biological Passport in Egypt Hanem A. Khalil a, Dina M. Abo-Elmatty b, Rosa V. Alemany c, Noha M. Mesbah b a Egyptian Anti-Doping Organization, Cairo, Egypt. b Faculty of Pharmacy, Department of Biochemistry Suez Canal University, Ismailia, Egypt. C Catalonian Anti-Doping Laboratory of Fundacio IMIM, Barcelona, Spain. Abstract Received on: 01.09. 2018 Using the Anabolic Androgenic Steroid (AAS) agents is evident not only Revised on: 21. 10. 2018 within the competitive senior and junior athletes, but also in non-sporting contexts by individuals seeking to „improve‟ their physique. No accurate data Accepted on: 01. 11. 2018 is available for the prevalence of AAS misuse among athletes. Studies suggest that it may be 1–5% of the population; with the prevalence being higher in males. Many studies documented side effects and health hazards with the misuse of anabolic steroids, where these were accused as a cause of Correspondence Author: deaths among athletes. Intake of exogenous anabolic steroids disturbed the Testosterone / Epitestosterone (T/E) ratio causing its evaluation above the Tel:+201270206648. normal level. This review outlines the anabolic steroids, its side effects and E-mail address: health impacts in both the sporting and physique development contexts. It also provides a brief review of the history of AAS as doping agents and [email protected] athlete biological passport. Conclusion: Doping among athletes is a widespread public health and social problem. Many studies have shown that both short- and long-term health complications have consequences and dependencies. -

Reproductive DHEA
Reproductive DHEA Analyte Information - 1 - DHEA Introduction DHEA (dehydroepiandrosterone), together with other important steroid hormones such as testosterone, DHT (dihydrotestosterone) and androstenedione, belongs to the group of androgens. Androgens are a group of C19 steroids that stimulate or control the development and maintenance of male characteristics. This includes the activity of the male sex organs and the development of secondary sex characteristics. Androgens are also precursors of all estrogens, the female sex hormones. DHEA (dehydroepiandrosterone) is the aromatic C19-steroid composed of a 10,13-dimethyl, 3-hydroxy group and 17-ketone. Its chemical name is 3β-hydroxy-5-androsten-17-one, its summary formula is C19H28O2, and its molecular weight (Mr) is 288.4 Da. The structural formulas of DHEA and related androgens are shown in Fig.1 Fig.1: Structural formulas of the most important androgens DHEA Androstenedione Testosterone Dihydrotestosterone There are more than 40 other names used for DHEA, including: (+)-Dehydroisoandrosterone; (3beta, 16alpha)-3,16-dihydroxy-androst-5-en- 17-one; 5,6-Dehydroisoandrosterone; 17-Chetovis, 17-Hormoforin, Andrestenol, Diandron, Prasterone and so on. As DHEA is very closely connected with its sulfate form DHEA-S, both hormones are mentioned together in the following text. Biosynthesis DHEA is the steroid hormone belonging to the weak androgens. DHEA and DHEA-S are the major C19 steroids produced from cholesterol by the zona reticularis of the adrenal cortex (Fig.2). DHEA is also produced in small quantities in the gonads (testis and ovary3,8,14), in adipose tissue and in the brain. From this point of view DHEA belongs to the neurosteroids22. -

Steroids and Other Appearance and Performance Enhancing Drugs (Apeds) Research Report
Research Report Revised Febrero 2018 Steroids and Other Appearance and Performance Enhancing Drugs (APEDs) Research Report Table of Contents Steroids and Other Appearance and Performance Enhancing Drugs (APEDs) Research Report Introduction What are the different types of APEDs? What is the history of anabolic steroid use? Who uses anabolic steroids? Why are anabolic steroids misused? How are anabolic steroids used? What are the side effects of anabolic steroid misuse? How does anabolic steroid misuse affect behavior? What are the risks of anabolic steroid use in teens? How do anabolic steroids work in the brain? Are anabolic steroids addictive? How are anabolic steroids tested in athletes? What can be done to prevent steroid misuse? What treatments are effective for anabolic steroid misuse? Where can I get further information about steroids? References Page 1 Steroids and Other Appearance and Performance Enhancing Drugs (APEDs) Research Report Esta publicación está disponible para su uso y puede ser reproducida, en su totalidad, sin pedir autorización al NIDA. Se agradece la citación de la fuente, de la siguiente manera: Fuente: Instituto Nacional sobre el Abuso de Drogas; Institutos Nacionales de la Salud; Departamento de Salud y Servicios Humanos de los Estados Unidos. Introduction Appearance and performance enhancing drugs (APEDs) are most often used by males to improve appearance by building muscle mass or to enhance athletic performance. Although they may directly and indirectly have effects on a user’s mood, they do not produce a euphoric high, which makes APEDs distinct from other drugs such as cocaine, heroin, and marijuana. However, users may develop a substance use disorder, defined as continued use despite adverse consequences. -

Michigan Department of Community Health
Michigan Department 0f Community Health, Office of Drug Control Policy The Michigan Association of Community Mental Health March 27, 2009 NationalNational TrendsTrends andand DeterrentDeterrent StrategiesStrategies ForFor PrescriptionPrescription andand OTCOTC DrugDrug AbuseAbuse Joseph Rannazzisi Deputy Assistant Administrator Office of Diversion Control Drug Enforcement Administration IntroductionIntroduction BackgroundBackground andand StatisticsStatistics RegulatoryRegulatory ControlControl MethodsMethods ofof DiversionDiversion InternetInternet DiversionDiversion CommonlyCommonly DivertedDiverted PharmaceuticalsPharmaceuticals Steroids/Steroids/hGHhGH DietaryDietary SupplementsSupplements SalviaSalvia DivinorumDivinorum TheThe 19601960’’ss Marijuana Seconal LSD Dexedrine Meprobamate TheThe 19701970’’ss Heroin T’s and Blues 4’s and Doors (Talwin and Pyrabenzamine) TheThe 19801980’’ss Tylenol w/Codeine and Doriden Hydromorphone Cocaine TheThe 19901990’’ss Oxycodone Methamphetamine 20002000 Hydrocodone Ketamine Alprazolam Flunitrazapam MDMA (Rohypnol) Scope and Extent of Problem 2004 2007 0.30.3 millionmillion 0.35 million SedativesSedatives 1.21.2 millionmillion 1.11.1 millionmillion StimulantsStimulants 1.61.6 millionmillion 1.81.8 millionmillion Anti-Anxiety Medication 4.44.4 millionmillion 5.25.2 millionmillion NarcoticNarcotic PainPain RelieversRelievers Source: 2004 and 2007 National Survey on Drug Use and Health TeensTeens andand TheirTheir AttitudesAttitudes 1 in 5 teens report abusing Rx medications to get high 2 in -
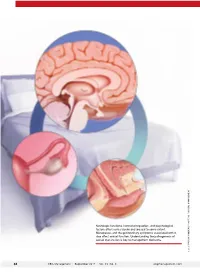
Neurologic Functions, Hormonal Regulation, and Psychological Factors Affect Sexual Desire and Arousal to Some Extent
Neurologic functions, hormonal regulation, and psychological factors affect sexual desire and arousal to some extent. Menopause, and the genitourinary symptoms associated with it, also affect sexual function. Understanding the pathogenesis of sexual dysfunction is key to management decisions. ILLUSTRATION: KIMBERLY MARTENS FOR OBG MANAGEMENT MARTENS KIMBERLY ILLUSTRATION: 34 OBG Management | September 2017 | Vol. 29 No. 9 obgmanagement.com UPDATE FEMALE SEXUAL DYSFUNCTION New and emerging treatment options hold promise for improving outcomes in this undertreated disorder ❯❯ Barbara S. Levy, MD Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC. The author reports no financial relationships relevant to this article. exual function is a complex, multifac- chronic conditions such as diabetes and S eted process mediated by neurologic back pain.4 functions, hormonal regulation, and psy- Understanding the pathogenesis of chological factors. What could possibly female sexual dysfunction helps to guide go wrong? our approach to its management. Indeed, IN THIS As it turns out, quite a lot. Female sex- increased understanding of its pathology has ARTICLE ual dysfunction is a common, vastly under- helped to usher in new and emerging treat- treated sexual health problem that can have ment options, as well as a personalized, bio- How hormones, wide-reaching effects on a woman’s life. psychosocial approach to its management. experience, These effects may include impaired body In this Update, I discuss the interplay of and behavior affect image, self-confidence, and self-worth. Sex- physiologic and psychological factors that the brain ual dysfunction also can contribute to rela- affect female sexual function as well as the page 36 tionship dissatisfaction and leave one feeling latest options for its management. -

Effect of Anabolic Steroids on the Cardiac and Skeletal Muscles of Adult Male Rats
International Journal of Clinical and Developmental Anatomy 2018; 4(1): 1-14 http://www.sciencepublishinggroup.com/j/ijcda doi: 10.11648/j.ijcda.20180401.11 ISSN: 2469-7990 (Print); ISSN: 2469-8008 (Online) Effect of Anabolic Steroids on the Cardiac and Skeletal Muscles of Adult Male Rats Hanan Abd-Elhakem Elgendy1, *, Adel Abd-Elmohdy Alhawary1, Mona Abd-Elrahim El-Shahat1, Afaf Taha Ali2 1Anatomy and Embryology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt 2Pathology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt Email address: *Corresponding author To cite this article: Hanan Abd-Elhakem Elgendy, Adel Abd-Elmohdy Alhawary, Mona Abd-Elrahim El-Shahat, Afaf Taha Ali. Effect of Anabolic Steroids on the Cardiac and Skeletal Muscles of Adult Male Rats. International Journal of Clinical and Developmental Anatomy. Vol. 4, No. 1, 2018, pp. 1-14. doi: 10.11648/j.ijcda.20180401.11 Received: November 28, 2017; Accepted: December 8, 2017; Published: February 7, 2018 Abstract: Abuse of anabolic androgenic steroids (AASs) by athletes has been increased rapidly in many countries to improve their physical fitness and appearance. The abuses of AASs have been associated with impacts on different systems of the body. The present study was conducted to evaluate the histological changes that occurred in skeletal and cardiac muscles during nandrolone (one of AASs) treatment histologically and immunohistochmically. Forty adult male albino rats were divided into four groups. Group 1; control group, group 2; was treated with nandrolone 5 mg/kg intramuscularly weekly, group 3 was treated with nandrolone10 mg/kg intramuscularly weekly and group 4; was treated with nandrolone 20 mg/kg intramuscularly weekly. -

Testosterone Prohormone Supplements
INVITED REVIEW Testosterone Prohormone Supplements GREGORY A. BROWN1, MATTHEW VUKOVICH2, and DOUGLAS S. KING3 1Human Performance Laboratory, University of Nebraska at Kearney, HPERLS Department, Kearney, NE; 2Human Performance Laboratory, South Dakota State University Department of HPER, Brooking, SD; and 3Exercise Biochemistry Laboratory, Department of Health and Human Performance, Iowa State University, Ames, IA ABSTRACT BROWN, G. A., M. VUKOVICH, and D. S. KING. Testosterone Prohormone Supplements. Med. Sci. Sports Exerc., Vol. 38, No. 8, pp. 1451–1461, 2006. Testosterone prohormones such as androstenedione, androstenediol, and dehydroepiandrosterone (DHEA) have been heavily marketed as testosterone-enhancing and muscle-building nutritional supplements for the past decade. Concerns over the safety of prohormone supplement use prompted the United States Food and Drug Administration to call for a ban on androstenedione sales, and Congress passed the Anabolic Steroid Control Act of 2004, which classifies androstenedione and 17 other steroids as controlled substances. As of January 2005, these substances cannot be sold without prescription. Here, we summarize the current scientific knowledge regarding the efficacy and safety of prohormone supplementation in humans. We focus primarily on androstenedione, but we also discuss DHEA, androstenediol, 19-nor androstenedione, and 19-nor androstenediol supplements. Contrary to marketing claims, research to date indicates that the use of prohormone nutritional supplements (DHEA, androstenedione, androstenediol, and other steroid hormone supplements) does not produce either anabolic or ergogenic effects in men. Moreover, the use of prohormone nutritional supplements may raise the risk for negative health consequences. Key Words: ANDROSTENEDIONE, ANDROSTENEDIOL, DHEA, NUTRITIONAL SUPPLEMENTS, TESTOSTERONE, ESTRADIOL ehydroepiandrosterone (DHEA), androstenedione, these compounds were not classified as anabolic steroids androstenediol, and similar testosterone precursor and could be purchased legally as dietary supplements. -

G. T'sjoen, Duprez D., Clement D
PSYCHO-ENDOCRINOLOGICAL ASPECTS IN PSYCHO-ENDOCRINOLOGICAL PERSONS MALES AND TRANSSEXUAL AGING Guy G. R. T’Sjoen Faculteit Geneeskunde & Gezondheidswetenschappen ACADEMISCH PROEFSCHRIFT PSYCHO-ENDOCRINOLOGICAL ASPECTS IN AGING MALES AND TRANSSEXUAL PERSONS ter verkrijging van de graad van doctor aan de Universiteit Gent, in het openbaar te verdedigen ten overstaan van de doctoraatscommissie van de Faculteit der Geneeskunde op vrijdag 20 januari 2006 om 17 uur in Auditorium C, Universtair Ziekenhuis Gent, De Pintelaan 185 door Guy T’Sjoen Geboren in Oudenaarde 26.07.1970 1 Promotor: Begeleidingscommissie: Prof. Dr. R. Rubens Prof. Dr. R. Rubens Prof. Dr. J. M. Kaufman Co-Promotor: Dr. G. De Cuypere Prof. Dr. J. M. Kaufman Examencommissie: Prof. Dr. J. Vande Walle (voorzitter) Prof. Dr. P. De Sutter Prof. Dr. L. Gooren Prof. Dr. P. Hoebeke Prof. Dr. J.J. Legros Prof. Dr. W. Oosterlinck Prof. Dr. D. Vanderschueren 2 Organon, Ipsen, Pfizer, GSK, Bayer, Novo Nordisk, Novartis, Sanofi, Aventis, Besins, Lilly, Menarini, Ellips and Sankyo kindly provided financial support for the publication of this thesis. Photographic illustrations by Dionisis Tsipiras, Mykonos, Greece. 3 Voor mijn ouders 4 “Una es más auténtica cuando más se parece a lo que ha soñado de sí misma...” Agrado in Pedro Almodóvar’s ‘Todo sobre mi Madre’. 5 CONTENTS Chapter 1 Aims of the Thesis and General Introduction ……..……………………… 9 PART I ……………………………………………………………………………….… 66 Chapter 2 Comparative assessment in young and elderly men of the gonadotropin response to aromatase inhibition …………………..…….. 67 Journal of Clinical Endocrinology & Metabolism 2005; 90: 5717-5722. Chapter 3 Perception of males’ aging symptoms, health and well-being in elderly com munity-dwelling men is not related to circulating androgen levels . -

2019 Prohibited List
THE WORLD ANTI-DOPING CODE INTERNATIONAL STANDARD PROHIBITED LIST JANUARY 2019 The official text of the Prohibited List shall be maintained by WADA and shall be published in English and French. In the event of any conflict between the English and French versions, the English version shall prevail. This List shall come into effect on 1 January 2019 SUBSTANCES & METHODS PROHIBITED AT ALL TIMES (IN- AND OUT-OF-COMPETITION) IN ACCORDANCE WITH ARTICLE 4.2.2 OF THE WORLD ANTI-DOPING CODE, ALL PROHIBITED SUBSTANCES SHALL BE CONSIDERED AS “SPECIFIED SUBSTANCES” EXCEPT SUBSTANCES IN CLASSES S1, S2, S4.4, S4.5, S6.A, AND PROHIBITED METHODS M1, M2 AND M3. PROHIBITED SUBSTANCES NON-APPROVED SUBSTANCES Mestanolone; S0 Mesterolone; Any pharmacological substance which is not Metandienone (17β-hydroxy-17α-methylandrosta-1,4-dien- addressed by any of the subsequent sections of the 3-one); List and with no current approval by any governmental Metenolone; regulatory health authority for human therapeutic use Methandriol; (e.g. drugs under pre-clinical or clinical development Methasterone (17β-hydroxy-2α,17α-dimethyl-5α- or discontinued, designer drugs, substances approved androstan-3-one); only for veterinary use) is prohibited at all times. Methyldienolone (17β-hydroxy-17α-methylestra-4,9-dien- 3-one); ANABOLIC AGENTS Methyl-1-testosterone (17β-hydroxy-17α-methyl-5α- S1 androst-1-en-3-one); Anabolic agents are prohibited. Methylnortestosterone (17β-hydroxy-17α-methylestr-4-en- 3-one); 1. ANABOLIC ANDROGENIC STEROIDS (AAS) Methyltestosterone; a. Exogenous* -

Identification in Vitro Des Métabolites De Nouveaux Stéroïdes : 17Α- Méthylstenbolone, 17Α-Méthylméthénolone Et Androst-2-En-17-One
Université du Québec INRS-Institut Armand-Frappier Identification in vitro des métabolites de nouveaux stéroïdes : 17α- méthylstenbolone, 17α-méthylméthénolone et androst-2-en-17-one Par Alexandre Sylvestre Mémoire ou thèse présentée pour l’obtention du grade de Maître es sciences (M.Sc.) en sciences expérimentales de la santé Jury d’évaluation Présidente du jury et Annie Castonguay examinatrice interne INRS-Institut Armand-Frappier Examinatrice externe Lekha Sleno Département de chimie Université du Québec à Montréal Directrice de recherche Christiane Ayotte INRS-Institut Armand-Frappier © Droits réservés de Alexandre Sylvestre, 2017 REMERCIEMENTS J’aimerais remercier Pre Christiane Ayotte de m’avoir fait confiance pour la réalisation d’un projet de maîtrise stimulant et enrichissant suite à deux stages effectués au laboratoire. Le partage de ses idées ainsi que ses connaissances tout au long du projet de recherche ont permis d’avoir de bons résultats à présenter. J’ai découvert un certain intérêt pour la recherche qui s’est développé au cours du projet ainsi que le plaisir de partager des connaissances. Je remercie toute l’équipe du laboratoire et les nombreux stagiaires (particulièrement Ashley et David) pour leur aide pendant ces deux années. Je tiens à souligner tous les bons moments passés au laboratoire ainsi que les multiples 5 à 7 qui m’ont permis d’apprécier mon travail. Je remercie Alain Charlebois pour son appui avec le fonctionnement des CG-SM ainsi que Alain Arseneault pour son aide essentielle avec le CLUHP. Toute l’équipe de la procédure « P4 » (Carolina, Julie, Marc-André, Marie, Natacha, Pauline, Philippe, Rita et Vicky) pour leur support et participation directe ou indirecte au projet.