Comprehensive Profiling of 8P11-12 Amplification in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

The Role of RNA Editing in Cancer Development and Metabolic Disorders
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 The oler of RNA editing in cancer development and metabolic disorders Che-Pei Kung Washington University School of Medicine in St. Louis Leonard B. Maggi Jr. Washington University School of Medicine in St. Louis Jason D. Weber Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Kung, Che-Pei; Maggi, Leonard B. Jr.; and Weber, Jason D., ,"The or le of RNA editing in cancer development and metabolic disorders." Frontiers in endocrinology.9,. 762. (2018). https://digitalcommons.wustl.edu/open_access_pubs/7400 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. REVIEW published: 18 December 2018 doi: 10.3389/fendo.2018.00762 The Role of RNA Editing in Cancer Development and Metabolic Disorders Che-Pei Kung 1,2*, Leonard B. Maggi Jr. 1,2 and Jason D. Weber 1,2,3* 1 ICCE Institute, Washington University School of Medicine, Saint Louis, MO, United States, 2 Division of Molecular Oncology, Department of Medicine, Washington University School of Medicine, Saint Louis, MO, United States, 3 Siteman Cancer Center, Department of Cell Biology and Physiology, Washington University School of Medicine, Saint Louis, MO, United States Numerous human diseases arise from alterations of genetic information, most notably DNA mutations. Thought to be merely the intermediate between DNA and protein, changes in RNA sequence were an afterthought until the discovery of RNA editing 30 years ago. -

Amplification of Chromosome 8 Genes in Lung Cancer Onur Baykara1, Burak Bakir1, Nur Buyru1, Kamil Kaynak2, Nejat Dalay3
Journal of Cancer 2015, Vol. 6 270 Ivyspring International Publisher Journal of Cancer 2015; 6(3): 270-275. doi: 10.7150/jca.10638 Research Paper Amplification of Chromosome 8 Genes in Lung Cancer Onur Baykara1, Burak Bakir1, Nur Buyru1, Kamil Kaynak2, Nejat Dalay3 1. Department of Medical Biology, Cerrahpasa Medical Faculty, Istanbul University, Turkey 2. Department of Chest Surgery, Cerrahpasa Medical Faculty, Istanbul University, Turkey 3. Department of Basic Oncology, I.U. Oncology Institute, Istanbul University, Turkey Corresponding author: Prof. Dr. Nejat Dalay, I.U.Oncology Institute, 34093 Capa, Istanbul, Turkey. e-mail : [email protected]; Phone : 90 542 2168861; Fax : 90 212 5348078 © 2015 Ivyspring International Publisher. Reproduction is permitted for personal, noncommercial use, provided that the article is in whole, unmodified, and properly cited. See http://ivyspring.com/terms for terms and conditions. Received: 2014.09.25; Accepted: 2014.12.18; Published: 2015.01.20 Abstract Chromosomal alterations are frequent events in lung carcinogenesis and usually display regions of focal amplification containing several overexpressed oncogenes. Although gains and losses of chromosomal loci have been reported copy number changes of the individual genes have not been analyzed in lung cancer. In this study 22 genes were analyzed by MLPA in tumors and matched normal tissue samples from 82 patients with non-small cell lung cancer. Gene amplifications were observed in 84% of the samples. Chromosome 8 was found to harbor the most frequent copy number alterations. The most frequently amplified genes were ZNF703, PRDM14 and MYC on chromosome 8 and the BIRC5 gene on chromosome 17. The frequency of deletions were much lower and the most frequently deleted gene was ADAM9. -

Mohammad Karbaschi Thesis
STRUCTURAL, PHYSIOLOGICAL AND MOLECULAR CHARACTERISATION OF THE AUSTRALIAN NATIVE RESURRECTION GRASS TRIPOGON LOLIIFORMIS (F.MUELL.) C.E.HUBB. DURING DEHYDRATION AND REHYDRATION Mohammad Reza Karbaschi Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Centre for Tropical Crops and Biocommodities Science and Engineering Faculty Queensland University of Technology November 2015 Keywords Arabidopsis thaliana; Agrobacterium-mediated transformation; Anatomy; Anti-apoptotic proteins; BAG4; Escherichia coli; Bulliform cells; C4 photosynthesis; Cell wall folding; Cell membrane integrity; Chaperone-mediated autophagy; Chlorophyll fluorescence; Hsc70/Hsp70; Desiccation tolerance, Dehydration; Drought; Electrolyte leakage; Freehand sectioning; Homoiochlorophyllous; Leaf structure; Leaf folding; Reactive oxygen species (ROS); Resurrection plant; Morphology; Monocotyledon; Nicotiana benthamiana; Photosynthesis; Physiology; Plant tissue; Programed cell death (PCD); Propidium iodide staining; Protein microarray chip; Sclerenchymatous tissue; Stress; Structure; Tripogon loliiformis; Ubiquitin; Vacuole fragmentation; Kranz anatomy; XyMS+; Structural, physiological and molecular characterisation of the Australian native resurrection grass Tripogon loliiformis (F.Muell.) C.E.Hubb. during dehydration and rehydration i Abstract Plants, as sessile organisms must continually adapt to environmental changes. Water deficit is one of the major environmental stresses that affects plants. While most plants can tolerate moderate dehydration -
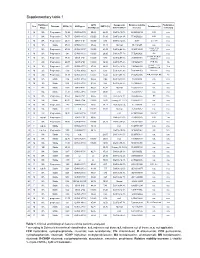
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Full Text (PDF)
Copyright © 2005 by the Genetics Society of America DOI: 10.1534/genetics.104.034322 Mutations in the Saccharomyces cerevisiae LSM1 Gene That Affect mRNA Decapping and 3 End Protection Sundaresan Tharun,*,1 Denise Muhlrad,† Ashis Chowdhury* and Roy Parker† *Department of Biochemistry, Uniformed Services University of the Health Sciences, Bethesda, Maryland 20814-4799 and †Department of Molecular and Cellular Biology and Howard Hughes Medical Institute, University of Arizona, Tucson, Arizona 85721 Manuscript received August 6, 2004 Accepted for publication January 20, 2005 ABSTRACT The decapping of eukaryotic mRNAs is a key step in their degradation. The heteroheptameric Lsm1p–7p complex is a general activator of decapping and also functions in protecting the 3Ј ends of deadenylated mRNAs from a 3Ј-trimming reaction. Lsm1p is the unique member of the Lsm1p–7p complex, distinguish- ing that complex from the functionally different Lsm2p–8p complex. To understand the function of Lsm1p, we constructed a series of deletion and point mutations of the LSM1 gene and examined their effects on phenotype. These studies revealed the following: (i) Mutations affecting the predicted RNA- binding and inter-subunit interaction residues of Lsm1p led to impairment of mRNA decay, suggesting that the integrity of the Lsm1p–7p complex and the ability of the Lsm1p–7p complex to interact with mRNA are important for mRNA decay function; (ii) mutations affecting the predicted RNA contact residues did not affect the localization of the Lsm1p–7p complex to the P-bodies; (iii) mRNA 3Ј-end protection could be indicative of the binding of the Lsm1p–7p complex to the mRNA prior to activation of decapping, since all the mutants defective in mRNA 3Ј end protection were also blocked in mRNA decay; and (iv) in addition to the Sm domain, the C-terminal domain of Lsm1p is also important for mRNA decay function. -

LSM1 Over-Expression in Saccharomyces Cerevisiae Depletes U6 Snrna Levels Natalie Luhtala1 and Roy Parker2,*
Published online 13 July 2009 Nucleic Acids Research, 2009, Vol. 37, No. 16 5529–5536 doi:10.1093/nar/gkp572 LSM1 over-expression in Saccharomyces cerevisiae depletes U6 snRNA levels Natalie Luhtala1 and Roy Parker2,* 1Cancer Biology Graduate Interdisciplinary Program and 2Department of Molecular and Cellular Biology & Howard Hughes Medical Institute, University of Arizona, Tucson, AZ, 85721-0106, USA Received May 19, 2009; Revised June 15, 2009; Accepted June 20, 2009 ABSTRACT trend analysis of the period from 2003 to 2005 revealed no significant changes in mortality rate (NCI SEER data- Lsm1 is a component of the Lsm1-7 complex base, http://seer.cancer.gov/statfacts/html/pancreas.html). involved in cytoplasmic mRNA degradation. Lsm1 Given that the current prognosis for patients with these is over-expressed in multiple tumor types, including tumors is dismal, it is vital that we search for novel ther- over 80% of pancreatic tumors, and increased levels apeutics targeting this disease. of Lsm1 protein have been shown to induce carcin- In 1997, Lsm1 was identified through subtractive ogenic effects. Therefore, understanding the per- hybridization cloning in pancreatic cancer cells (1) and turbations in cell process due to increased Lsm1 was shown to be over-expressed in 87% of pancreatic protein may help to identify possible therapeutics cancers. Subsequently, its over-expression has been targeting tumors over-expressing Lsm1. Herein, we described in 40% of prostate cancers (2), a subset show that LSM1 over-expression in the yeast (15–20%) of breast cancers that are amplified at the Saccharomyces cerevisiae inhibits growth primarily 8p11-12 region (3,4) and most recently in lung cancers due to U6 snRNA depletion, thereby altering pre- and mesotheliomas (5). -

Cellular and Molecular Investigations of Undiagnosed Neurometabolic
CELLULAR AND MOLECULAR INVESTIGATIONS OF UNDIAGNOSED NEUROMETABOLIC DISORDERS Submitted in application for award of Doctor of Philosophy (PhD) Emma Reid Centre for Translational Omics Genetics and Genomic Medicine Institute of Child Health University College London August 2016 DECLARATION I, Emma Reid, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Where experimental or analytical work has been completed by others, this has been stated in the relevant section of this thesis. However, these instances are also listed below: • Alignment of sequencing reads to the reference genome, variant calling (Section 2.4.11) and ExomeDepth analysis (Section 3.3.3) of the gene panel sequencing data was performed by Dr Chris Boustred (NE Thames Regional Genetics Service, GOSH, UK). • Whole exome sequencing of patients X, 1, 4 and 5 was outsourced to BGI Genomics Hong Kong and the resulting raw data files were processed and aligned by Dr Chela James (GOSgene, ICH, UK) (Section 2.5). • Whole exome sequencing of patient Y and processing of the resulting raw data was performed by Dr Olaf Bodamer (University of Miami, USA) (Section 6.4.1). • Homozygosity mapping and whole exome sequencing of index PROSC family was performed by Dr Niklas Darin (The Queen Silvia Children’s Hospital, Sweden) (Section 7.1.4). • Sanger sequencing of additional PROSC and PNPO-deficient patients was performed by Dr Philippa Mills (ICH, UK) (Section 7.2). • Sample processing for electron microscopy analysis was carried out by Elizabeth Latimer- Bowman (Histopathology Department, GOSH, UK) and imaging was performed by Glenn Anderson (Histopathology Department, GOSH, UK) (Section 2.14.2). -

Metastatic Adrenocortical Carcinoma Displays Higher Mutation Rate and Tumor Heterogeneity Than Primary Tumors
ARTICLE DOI: 10.1038/s41467-018-06366-z OPEN Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors Sudheer Kumar Gara1, Justin Lack2, Lisa Zhang1, Emerson Harris1, Margaret Cam2 & Electron Kebebew1,3 Adrenocortical cancer (ACC) is a rare cancer with poor prognosis and high mortality due to metastatic disease. All reported genetic alterations have been in primary ACC, and it is 1234567890():,; unknown if there is molecular heterogeneity in ACC. Here, we report the genetic changes associated with metastatic ACC compared to primary ACCs and tumor heterogeneity. We performed whole-exome sequencing of 33 metastatic tumors. The overall mutation rate (per megabase) in metastatic tumors was 2.8-fold higher than primary ACC tumor samples. We found tumor heterogeneity among different metastatic sites in ACC and discovered recurrent mutations in several novel genes. We observed 37–57% overlap in genes that are mutated among different metastatic sites within the same patient. We also identified new therapeutic targets in recurrent and metastatic ACC not previously described in primary ACCs. 1 Endocrine Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA. 2 Center for Cancer Research, Collaborative Bioinformatics Resource, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA. 3 Department of Surgery and Stanford Cancer Institute, Stanford University, Stanford, CA 94305, USA. Correspondence and requests for materials should be addressed to E.K. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:4172 | DOI: 10.1038/s41467-018-06366-z | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-06366-z drenocortical carcinoma (ACC) is a rare malignancy with types including primary ACC from the TCGA to understand our A0.7–2 cases per million per year1,2. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing