The Meaning of Time in Quantum Mechanics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

User Manual for Amazfit GTR 2 (English Edition) Contents
User Manual for Amazfit GTR 2 (English Edition) Contents User Manual for Amazfit GTR 2 (English Edition) ......................................................................................1 Getting started................................................................................................................................................3 Appearance ....................................................................................................................................3 Power on and off............................................................................................................................3 Charging ........................................................................................................................................3 Wearing & Replacing Watch Strap ...............................................................................................4 Connecting & Pairing ....................................................................................................................4 Updating the system of your watch ...............................................................................................5 Control center ................................................................................................................................5 Time System..................................................................................................................................6 Units...............................................................................................................................................6 -

SOFA Time Scale and Calendar Tools
International Astronomical Union Standards Of Fundamental Astronomy SOFA Time Scale and Calendar Tools Software version 1 Document revision 1.0 Version for Fortran programming language http://www.iausofa.org 2010 August 27 SOFA BOARD MEMBERS John Bangert United States Naval Observatory Mark Calabretta Australia Telescope National Facility Anne-Marie Gontier Paris Observatory George Hobbs Australia Telescope National Facility Catherine Hohenkerk Her Majesty's Nautical Almanac Office Wen-Jing Jin Shanghai Observatory Zinovy Malkin Pulkovo Observatory, St Petersburg Dennis McCarthy United States Naval Observatory Jeffrey Percival University of Wisconsin Patrick Wallace Rutherford Appleton Laboratory ⃝c Copyright 2010 International Astronomical Union. All Rights Reserved. Reproduction, adaptation, or translation without prior written permission is prohibited, except as al- lowed under the copyright laws. CONTENTS iii Contents 1 Preliminaries 1 1.1 Introduction ....................................... 1 1.2 Quick start ....................................... 1 1.3 The SOFA time and date routines .......................... 1 1.4 Intended audience ................................... 2 1.5 A simple example: UTC to TT ............................ 2 1.6 Abbreviations ...................................... 3 2 Times and dates 4 2.1 Timekeeping basics ................................... 4 2.2 Formatting conventions ................................ 4 2.3 Julian date ....................................... 5 2.4 Besselian and Julian epochs ............................. -

The Matter of Time
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 15 June 2021 doi:10.20944/preprints202106.0417.v1 Article The matter of time Arto Annila 1,* 1 Department of Physics, University of Helsinki; [email protected] * Correspondence: [email protected]; Tel.: (+358 44 204 7324) Abstract: About a century ago, in the spirit of ancient atomism, the quantum of light was renamed the photon to suggest its primacy as the fundamental element of everything. Since the photon carries energy in its period of time, a flux of photons inexorably embodies a flow of time. Time comprises periods as a trek comprises legs. The flows of quanta naturally select optimal paths, i.e., geodesics, to level out energy differences in the least time. While the flow equation can be written, it cannot be solved because the flows affect their driving forces, affecting the flows, and so on. As the forces, i.e., causes, and changes in motions, i.e., consequences, cannot be separated, the future remains unpre- dictable, however not all arbitrary but bounded by free energy. Eventually, when the system has attained a stationary state, where forces tally, there are no causes and no consequences. Then time does not advance as the quanta only orbit on and on. Keywords: arrow of time; causality; change; force; free energy; natural selection; nondeterminism; quantum; period; photon 1. Introduction We experience time passing, but the experience itself lacks a theoretical formulation. Thus, time is a big problem for physicists [1-3]. Although every process involves a passage of time, the laws of physics for particles, as we know them today, do not make a difference whether time flows from the past to the future or from the future to the past. -

Changing the Time on Your Qqest Time Clock For
IntelliClockDaylight SavingSeries Time CHANGING THE TIME ON YOUR QQEST IQTIME 1000 CLOCK FOR DAYLIGHT SAVING TIME HardwareIt’s Daylight Specifications Saving Time Automatic DST Uploads main ClockLink screen. Highlight the clock that you would like to upload again! Make sure that your the date and time to and click on the clocks are reset before the The ClockLink Scheduler contains a “Connect” link. The ClockLink utility time changes. pre-configured Daylight Saving script connects to the selected time clock. As our flagship data collection terminal, Dimensionsintended & Weight: to automatically upload the 8.75” x 8.25” x 1.5”, approx. 1.65 lbs.; with the IQ1000, our most advanced time time to your clocks each Daylight . Once you have connected to a time biometric authentication module 1.9 lbs HID Proximity3 Card Reader: clock, delivers the capabilities required Saving. This script already exists in clock, the clock options are displayed the ClockLink Scheduler, and cannot HID 26 bit and 37 bit formats supported for even the most demanding Keypad: 25 keys (1-9, (decimal point), CLEAR, on the right-hand side of the screen. be edited or deleted. Since Windows The row of icons at the top of the applications. ENTER,automatically MENU, Lunch updates and Meal/Br the eakcomputer’s Keys, Communication Options: Job Costing and Tracking Keys, Department Directscreen Ethernet allows or Cellular you to select which time for Daylight Saving, the time offset functions you would like to perform at Transfernever Keys, needs Tip/Gratuity to be updated.Keys. Key “Click”. Selectable by user. If enabled, the clock will Cellular:the GSM clock. -

Clock/Calendar Implementation on the Stm32f10xxx Microcontroller RTC
AN2821 Application note Clock/calendar implementation on the STM32F10xxx microcontroller RTC The internal RTC (real-time clock) of the 32-bit, ARM® Cortex™-M3-based medium-density STM32F10xxx microcontroller is an independent timer. It provides a set of continuously running counters, which, using suitable software, can be used to implement a clock/calendar function. The RTC and backup registers are supplied through a switch that draws power from either the VDD supply (when present) or the backup battery. Thus the RTC runs even when the main supply is switched off. This application note gives an example of how to implement the clock/calendar functionality using the medium-density STM32F10xxx microcontroller in low-power and standard applications. The firmware example also performs leap year counting and tamper time stamping. It provides a low-power mode demonstration and a basic RTC calibration routine to compensate for crystal and temperature variations. The STM3210B-EVAL board supports the firmware example described in this application note without any modification to its hardware. Figure 1. STM3210B-EVAL board April 2009 Doc ID 14949 Rev 2 1/28 www.st.com Contents AN2821 Contents 1 Overview of the medium-density STM32F10xxx backup domain . 6 1.1 Main backup domain features . 6 1.2 Main RTC features . 7 2 Configuring the RTC registers . 8 3 Clock/calendar functionality features . 9 3.1 Clock/calendar basics . 9 3.1.1 Implementing the clock function on the medium-density STM32F10xxx . 9 3.1.2 Implementing calendar function on the medium-density STM32F10xxx . 9 3.1.3 Summer time correction . 11 3.2 Clock source selection . -

Automatically Setting Daylight Saving Time on MISER
DST SETTINGS Effective: Revision: 12/01/10 Draft Automa tically Setting Daylight Saving Time on MISER NTP The Network Time Protocol (NTP) provides synchronized timekeeping among a set of distributed time servers and clients. The local OpenVMS host maintains an NTP configuration file ( TCPIP$NTP.CONF) of participating peers. Before configuring your host, you must do the following: 1) Select time sources. 2) Obtain the IP addresses or host names of the time sources. 3) Enable the NTP service via the TCPIP$CONFIG command procedure. TCPIP$NTP.CONF is maintained in the SYS$SPECIFIC[TCPIP$NTP] directory. NOTE: The NTP configuration file is not dynamic; it requires restarting NTP after editing for the changes to take effect. NTP Startup and Shutdown To stop and then restart the NTP server to allow configuration changes to take effect, you must execute the following commands from DCL: $ @sys$startup:tcpip$ntp_shutdown $ @sys$startup:tcpip$ntp_startup NOTE: Since NTP is an OpenVMS TCP/IP service, there is no need to modify any startup command procedures to include these commands; normal TCP/IP startup processing will check to see if it should start or stop the service at the right time. NTP Configuration File Statements Your NTP configuration file should always include the following driftfile entry. driftfile SYS$SPECIFIC:[TCPIP$NTP]TCPIP$NTP.DRIFT CONFIDENTIAL All information contained in this document is confidential and is the sole property of HSQ Technology. Any reproduction in part or whole without the written permission of HSQ Technology is prohibited. PAGE: 1 of 9 DST SETTINGS ] NOTE: The driftfile is the name of the file that stores the clock drift (also known as frequency error) of the system clock. -

Outbound Contact Deployment Guide
Outbound Contact Deployment Guide Time Zones 9/30/2021 Time Zones Time Zones Contents • 1 Time Zones • 1.1 Creating a Custom Time Zone for a Tenant • 1.2 Time Zones Object--Configuration Tab Fields • 1.3 Time Zones and Time-Related Calculations Outbound Contact Deployment Guide 2 Time Zones Outbound Contact uses time zones in call records to determine the contact's time zone. Genesys Administrator populates the tz_dbid field with the international three-letter abbreviation for the time zone parameter when it imports or exports a calling list. Call time, dial schedule time, and valid dial time (dial from and dial till) are based on the record's time-zone. For more information about the time zone abbreviations see Framework Genesys Administrator Help. If Daylight Savings Time (DST) is configured for time zones located below the equator using the Current Year or Fixed Date (Local) properties, define Note: both the Start Date and End Date in the DST definition as the current year and make the Start Date later than the End Date. Outbound Contact dynamically updates time changes from winter to summer and summer to winter. The default set of Time Zones created during Configuration Server installation is located in the Time Zones folder under the Environment (for a multi-tenant environment) or under the Resources folder (for a single tenant). Creating a Custom Time Zone for a Tenant • To create a custom Time Zone for a Tenant, if necessary. Start 1. In Genesys Administrator, select Provisioning > Environment > Time Zones. 2. Click New. 3. Define the fields in the Configuration tab. -

IBM Often Gets the Question About What to Do Regarding Leap Seconds in the Mainframe Environment
Leap Seconds and Server Time Protocol (STP) What should I do? There is plenty of documentation in the Server Time Protocol Planning Guide and Server Time Protocol Implementation Guide regarding Leap Seconds. This white paper will not attempt to rehash the fine-spun detail that is already written there. Also, this paper will not try to explain why leap seconds exist. There is plenty of information about that on-line. See http://en.wikipedia.org/wiki/Leap_second for a reasonable description of leap seconds. Leap seconds are typically inserted into the international time keeping standard either at the end of June or the end of December, if at all. Leap second adjustments occur at the same time worldwide. Therefore, it must be understood when UTC 23:59:59 will occur in your particular time zone, because this is when a scheduled leap second offset adjustment will be applied. Should I care? IBM often gets the question about what to do regarding leap seconds in the mainframe environment. As usual, the answer “depends”. You need to decide what category you fit into. 1. My server needs to be accurate, the very instant a new leap second occurs. Not many mainframe servers or distributed servers fit into this category. Usually, if you fit into this category, you already know it. 2. My server needs to be accurate, within one second or so. I can live with it, as long as accurate time is gradually corrected (steered) by accessing an external time source (ETS). This is more typical for most mainframe users. Category 1 If you fit into category 1, there are steps that need to be taken. -

System Time/NTP/PTP Configuration Module V1.0
System Time/NTP/PTP configuration module v1.0 Copyright © Riverbed Technology Inc. 2018 Created Sep 23, 2020 at 05:09 PM Resource: current_time Current system time http://{device}/api/mgmt.time/1.0/now JSON { "is_dst": boolean, "timestamp": integer, "tz_name": string, "utc_offset": integer } Property Name Type Description Notes current_time <object> Current system time current_time.is_dst <boolean> Daylight saving status Read-only; Optional; current_time.timestamp <integer> Current system time, in Unix epoch seconds Optional; current_time.tz_name <string> Time zone abbreviation, e.g., PST Read-only; Optional; current_time.utc_offset <integer> The UTC offset in seconds Read-only; Optional; Links current_time: get GET http://{device}/api/mgmt.time/1.0/now Response Body Returns a current_time data object. current_time: set PUT http://{device}/api/mgmt.time/1.0/now Request Body Provide a current_time data object. Response Body Returns a current_time data object. Resource: ntp_server An NTP server http://{device}/api/mgmt.time/1.0/ntp/servers/items/{server_id} JSON { "address": string, "encryption": string, "key_id": integer, "prefer": boolean, "secret": string, "server_id": integer, "version": integer } Property Name Type Description Notes ntp_server <object> An NTP server ntp_server.address <string> NTP server address Optional; ntp_server.encryption <string> Encryption method to use Optional; Values: none, sha1, md5; ntp_server.key_id <integer> Encryption key ID Optional; Range: 0 to 65534; ntp_server.prefer <boolean> Prefer this server Optional; -

Change Without Time Relationalism and Field Quantization
Change Without Time Relationalism and Field Quantization Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) der naturwissenschaftlichen Fakult¨at II – Physik der Universit¨at Regensburg vorgelegt von Johannes Simon aus Schwandorf 2004 Promotionsgesuch eingereicht am: 19.04.2004 Die Arbeit wurde angeleitet von Prof. Dr. Gustav M. Obermair. Prufungsausschuss:¨ Vorsitzender Prof. Dr. W. Prettl 1. Gutachter Prof. Dr. G. M. Obermair 2. Gutachter Prof. Dr. E. Werner weiterer Prufer¨ Prof. Dr. A. Sch¨afer Tag der mundlichen¨ Prufung:¨ 23.07.2004 Tempus item per se non est [...] Titus Lucretius Carus [Car, 459 ff] Preface This thesis studies the foundations of time in physics. Its origin lies in the urge to understand the quantum measurement problem. While the emergence of classical- ity can be well described within algebraic quantum mechanics of infinite systems, this can be achieved only in infinite time. This led me to a study of quantum dy- namics of infinite systems, which turned out to be far less unique than in the case of finitely many degrees of freedom. In deciding on the correct time evolution the question appears how time – or rather duration – is being measured. Traditional quantum mechanics lacks a time observable, and for closed systems in an energy eigenstate duration is indeed meaningless. A similar phenomenon shows up in general relativity, where absolute duration (as well as spatial distance) becomes meaningless due to diffeomorphism invariance. However, by relating different parts of a closed system through simultaneity (in quantum mechanics as well as in general relativity), an internal notion of time becomes meaningful. This similarity between quantum mechanics and general relativity was recognized in the context of quantum gravity by Carlo Rovelli, who proposed a relational concept of quantum time in 1990. -
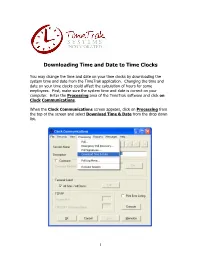
Downloading Time and Date to Time Clocks
Downloading Time and Date to Time Clocks You may change the time and date on your time clocks by downloading the system time and date from the TimeTrak application. Changing the time and date on your time clocks could affect the calculation of hours for some employees. First, make sure the system time and date is correct on your computer. Enter the Processing area of the TimeTrak software and click on Clock Communications. When the Clock Communications screen appears, click on Processing from the top of the screen and select Download Time & Date from the drop down list. 1 In the next screen to appear, you may choose to download time and date to all clocks or select individual clocks. From the Terminal Select & Proceed screen, click the Edit button under Terminal Select. 2 The Select Items screen will appear. All of the time clocks connected to the TimeTrak system will be listed by Site Number, Clock ID, and Description. Place a check mark in the box next to each clock you wish to download time and date. Then click Accept. 3 The Terminal Select & Proceed screen will return. Upon selecting the Proceed button, the TimeTrak application will begin a communication session with the time clocks and download the time and date. When the communication session has completed, the process will return to the Clock Communications screen. You may then click OK or Cancel to return to TimeTrak’s main screen. More helpful information may be found at www.timeclocksales.com 4. -

Fundamentals on UTC Relationship Between GNSS Times
Bureau International des Poids et Mesures Fundamentals on UTC Relationship between GNSS times E.F. Arias and W. Lewandowski 4th ICG, 14-18 September 2009, St Petersburg Bureau International des Poids et Mesures Outline of presentation ••• Definition of international time scales ••• UTC ••• TAI ••• Leap second ••• Relation between satellite time scales ••• GPS time ••• Glonass time ••• Galileo system time ••• COMPASS system time … Bureau International des Poids et Mesures Unification of time 1884 - Adoption of a prime meridian Greenwich and of an associated time - universal time, based on the rotation of the Earth. 1948 - International Astronomical Union recommends the use of Universal Time (UT). 1968 - 13th General Conference of Weights and Measures adopted a definition of SI second, based on a caesium transition, and opened the way toward the formal definition of International Atomic Time (TAI). 1971 - International Astronomical Union, International Telecommunications Union, General Conference of Weights and Measures recommend the use of Coordinated Universal Time (UTC) based on TAI. Introduction of leap seconds. 2003 - Use of leap seconds under revision Bureau International des Poids et Mesures Coordinated Universal Time (UTC) UTC is computed at the BIPM and made available every month in the BIPM Circular T through the publication of [UTC – UTC(k)] ] International Atomic Time (TAI) is based on the readings of about 400 atomic clocks located in metrology institutes in about 45 countries around the world. TAI has scientific applications