Impact of Genetics on Meat Quality of Pigs and Beef Cattle by Huaigang Lei a Thesis Submitted in Partial Fulfillment of the Requ
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ERK Phosphorylation of MED14 in Promoter Complexes During Mitogen-Induced Gene Activation by Elk-1 Matthew D
Published online 17 September 2013 Nucleic Acids Research, 2013, Vol. 41, No. 22 10241–10253 doi:10.1093/nar/gkt837 ERK phosphorylation of MED14 in promoter complexes during mitogen-induced gene activation by Elk-1 Matthew D. Galbraith1,2, Janice Saxton1,LiLi1, Samuel J. Shelton1,3, Hongmei Zhang1,4, Joaquin M. Espinosa2 and Peter E. Shaw1,* 1School of Biomedical Sciences, University of Nottingham, Queen’s Medical Centre, Nottingham, NG7 2UH, UK, 2Department of Molecular, Cellular and Developmental Biology, Howard Hughes Medical Institute, University of Colorado at Boulder, Boulder, CO 80309, USA, 3Department of Neurology, Center of Regeneration Medicine and Stem Cell Research, University of California, San Francisco, CA 90089, USA and 4Department of Pharmacology, Ningxia Medical University, Yinchuan 750004, China Received March 22, 2013; Revised August 21, 2013; Accepted August 28, 2013 ABSTRACT INTRODUCTION The ETS domain transcription factor Elk-1 stimu- The specific and temporal co-ordination of gene expres- lates expression of immediate early genes (IEGs) in sion is a fundamental process of cell-based life with response to mitogens. These events require phos- patterns of gene expression controlling proliferation, dif- phorylation of Elk-1 by extracellular signal-regulated ferentiation and cell death. Immediate early gene (IEG) expression has revealed how crucial regulatory events kinase (ERK) and phosphorylation-dependent inter- revolve around the interface between pathway-specific action of Elk-1 with co-activators, including histone transcription factors and components of the transcription acetyltransferases and the Mediator complex. Elk-1 machinery, involving a plethora of protein interactions also recruits ERK to the promoters of its target and modifications (1,2). -

Downloaded on November Tured Output Support Vector Machine (SVM) Approach 6, 2013
Funk et al. Journal of Biomedical Semantics (2015) 6:9 DOI 10.1186/s13326-015-0006-4 JOURNAL OF BIOMEDICAL SEMANTICS RESEARCH Open Access Evaluating a variety of text-mined features for automatic protein function prediction with GOstruct Christopher S Funk1*, Indika Kahanda2, Asa Ben-Hur2 and Karin M Verspoor3,4 Abstract Most computational methods that predict protein function do not take advantage of the large amount of information contained in the biomedical literature. In this work we evaluate both ontology term co-mention and bag-of-words features mined from the biomedical literature and analyze their impact in the context of a structured output support vector machine model, GOstruct. We find that even simple literature based features are useful for predicting human protein function (F-max: Molecular Function = 0.408, Biological Process = 0.461, Cellular Component = 0.608). One advantage of using literature features is their ability to offer easy verification of automated predictions. We find through manual inspection of misclassifications that some false positive predictions could be biologically valid predictions based upon support extracted from the literature. Additionally, we present a “medium-throughput” pipeline that was used to annotate a large subset of co-mentions; we suggest that this strategy could help to speed up the rate at which proteins are curated. Keywords: Text mining, Protein function prediction, Biomedical concept recognition Introduction Background Characterizing the functions of proteins is an important Literature mining has been shown to have substantial task in bioinformatics today. In recent years, many com- promise in the context of automated function prediction, putational methods to predict protein function have been although there has been limited exploration to date [2]. -

Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo)
animals Article Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo) Kyu-Sang Lim 1 , Sun-Sik Chang 2, Bong-Hwan Choi 3, Seung-Hwan Lee 4, Kyung-Tai Lee 3 , Han-Ha Chai 3, Jong-Eun Park 3 , Woncheoul Park 3 and Dajeong Lim 3,* 1 Department of Animal Science, Iowa State University, Ames, IA 50011, USA; [email protected] 2 Hanwoo Research Institute, National Institute of Animal Science, Rural Development Administration, Pyeongchang 25340, Korea; [email protected] 3 Animal Genomics and Bioinformatics Division, National Institute of Animal Science, Rural Development Administration, Wanju 55365, Korea; [email protected] (B.-H.C.); [email protected] (K.-T.L.); [email protected] (H.-H.C.); [email protected] (J.-E.P.); [email protected] (W.P.) 4 Division of Animal and Dairy Science, Chungnam National University, Daejeon 34134, Korea; [email protected] * Correspondence: [email protected] Received: 26 July 2019; Accepted: 23 September 2019; Published: 26 September 2019 Simple Summary: Allele-specific expression (ASE) is the biased allelic expression of genetic variants within the gene. Recently, the next-generation sequencing (NGS) technologies allowed us to detect ASE genes at a transcriptome-wide level. It is essential for the understanding of animal development, cellular programming, and the effect on their complexity because ASE shows developmental, tissue, or species-specific patterns. However, these aspects of ASE still have not been annotated well in farm animals and most studies were conducted mainly at the fetal stages. Hence, the current study focuses on detecting ASE genes in 17 tissues in adult cattle. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle
Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, Mekki Boussaha, Fabio Pilla, Baldassare Portolano, Salvatore Mastrangelo To cite this version: Slim Ben-Jemaa, Gabriele Senczuk, Elena Ciani, Roberta Ciampolini, Gennaro Catillo, et al.. Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle. Frontiers in Genetics, Frontiers, 2021, 10.3389/fgene.2021.675569. hal-03210766 HAL Id: hal-03210766 https://hal.inrae.fr/hal-03210766 Submitted on 28 Apr 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ORIGINAL RESEARCH published: 28 April 2021 doi: 10.3389/fgene.2021.675569 Genome-Wide Analysis Reveals Selection Signatures Involved in Meat Traits and Local Adaptation in Semi-Feral Maremmana Cattle Slim Ben-Jemaa 1, Gabriele Senczuk 2, Elena Ciani 3, Roberta -

University of Leicester, Msc Medical Statistics, Thesis, Wilmar
Thesis MSc in Medical Statistics Department of Health Sciences University of Leicester, United Kingdom Application of Bayesian hierarchical generalized linear models using weakly informative prior distributions to identify rare genetic variant effects on blood pressure Wilmar Igl March 2015 Summary Background Currently rare genetic variants are discussed as a source of \missing heritability" of complex traits. Bayesian hierarchical models were proposed as an efficient method for the estimation and aggregation of conditional effects of rare variants. Here, such models are applied to identify rare variant effects on blood pressure. Methods Empirical data provided by the Genetic Analysis Workshop 19 (2014) included 1,851 Mexican-American individuals with diastolic blood pressure (DBP), systolic blood pressure (SBP), hypertension (HTN) and 461,868 variants from whole- exome sequencing of odd-numbered chromosomes. Bayesian hierarchical generalized linear models using weakly informative prior distributions were applied. Results Associations of rare variants chr1:204214013 (estimate = 39.6, Credible In- terval (CrI) 95% = [25.3, 53.9], Bayesian p = 6:8 × 10−8) in the PLEKHA6 gene and chr11:118518698 (estimate = 32.2, CrI95% = [20.6, 43.9], Bayesian p = 7:0 × 10−8) in the PHLEDB1 gene were identified. Joint effects of grouped rare variants on DBP in 23 genes (Bayesian p = [8:8 × 10−14, 9:3 × 10−8]) and on SBP in 21 genes (Bayesian p = [8:6 × 10−12, 7:8 × 10−8]) in pathways related to hemostasis, sodi- um/calcium transport, ciliary activity, and others were found. No association with hypertension was detected. Conclusions Bayesian hierarchical generalized linear models with weakly informa- tive priors can successfully be applied in exome-wide genetic association analyses of rare variants. -
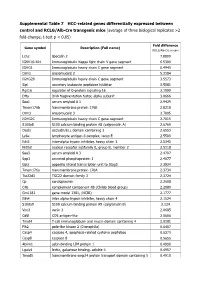
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

UC Riverside UC Riverside Previously Published Works
UC Riverside UC Riverside Previously Published Works Title Analysis of the Candida albicans Phosphoproteome. Permalink https://escholarship.org/uc/item/0663w2hr Journal Eukaryotic cell, 14(5) ISSN 1535-9778 Authors Willger, SD Liu, Z Olarte, RA et al. Publication Date 2015-05-01 DOI 10.1128/ec.00011-15 License https://creativecommons.org/licenses/by-nc-sa/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Analysis of the Candida albicans Phosphoproteome S. D. Willger,a Z. Liu,b R. A. Olarte,c M. E. Adamo,d J. E. Stajich,c L. C. Myers,b A. N. Kettenbach,b,d D. A. Hogana Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire, USAa; Department of Biochemistry, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire, USAb; Department of Plant Pathology and Microbiology, University of California, Riverside, California, USAc; Norris Cotton Cancer Center, Geisel School of Medicine at Dartmouth, Lebanon, New Hampshire, USAd Candida albicans is an important human fungal pathogen in both immunocompetent and immunocompromised individuals. C. albicans regulation has been studied in many contexts, including morphological transitions, mating competence, biofilm forma- tion, stress resistance, and cell wall synthesis. Analysis of kinase- and phosphatase-deficient mutants has made it clear that pro- tein phosphorylation plays an important role in the regulation of these pathways. In this study, to further our understanding of phosphorylation in C. albicans regulation, we performed a deep analysis of the phosphoproteome in C. albicans. We identified 19,590 unique peptides that corresponded to 15,906 unique phosphosites on 2,896 proteins. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 During Serum-Starvation-Induced Ciliogenesis
cells Article SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis Pengtao Wang 1,2,3,4, Jianhong Xia 2,3, Leilei Zhang 2,3, Shaoyang Zhao 2,3, Shengbiao Li 2,3, Haiyun Wang 2,3, Shan Cheng 4, Heying Li 2,3, Wenguang Yin 2,3, Duanqing Pei 2,3,5,* and Xiaodong Shu 2,3,4,5,6,* 1 School of Life Sciences, University of Science and Technology of China, Hefei 230027, China; [email protected] 2 CAS Key Laboratory of Regenerative Biology, South China Institutes for Stem Cell Biology and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China; [email protected] (J.X.); [email protected] (L.Z.); [email protected] (S.Z.); [email protected] (S.L.); [email protected] (H.W.); [email protected] (H.L.); [email protected] (W.Y.) 3 Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine, Guangzhou 510530, China 4 Hefei Institute of Stem Cell and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China; [email protected] 5 Guangzhou Regenerative Medicine and Health Guangdong Laboratory (GRMH-GDL), Guangzhou 510005, China 6 Joint School of Life Sciences, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou Medical University, Guangzhou 511436, China * Correspondence: [email protected] (D.P.); [email protected] (X.S.); Tel.: +86-20-3201-5310 (X.S.) Academic Editor: Tomer Avidor-Reiss Received: 13 September 2019; Accepted: 27 October 2019; Published: 29 October 2019 Abstract: Centriolar satellites are non-membrane cytoplasmic granules that deliver proteins to centrosome during centrosome biogenesis and ciliogenesis.