And Stanna-Closo-Dodecaborate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chem-601: Structure and Bonding of Molecules and Materials
Chem-601: Structure and Bonding of Molecules and Materials Instructor: Efrain E. Rodriguez [email protected] Office: Chemistry 4101 Office hours: MW 2-3, and by appointment Lectures: Chemistry 0122 MWF, 10 AM – 10:50 AM Grading scheme: 8 problem sets 150 points total Midterm exams (2) 75 points each Final exam 100 points TOTAL 400 points Textbooks: 1) G L. Miessler and D. A. Tarr, Inorganic Chemistry, 4th Ed., Pearson, 2011. 2) R. L. Carter, Molecular Symmetry and Group Theory, Wiley, 1998. Tentative lecture schedule (readings in parentheses): 1.) A survey of inorganic and materials chemistry today 2.) Atomic structure a. Refresher on quantum and hydrogen-like orbitals (IC 2.1 to 2.2.2). b. Multi-electron orbitals, aufbau principle, and periodicity (IC rest of Ch. 2). 3.) Simple bonding models in molecules and solids a. Octet rule, VSPER structures, the valence bond model and resonance (IC Ch 3). b. Ionic bonding, prototype solid structures, Born-Haber cycle to calculate lattice energies (IC 7.1 to 7.2.2 ). 4.) Group theory a. Symmetry elements, point groups, and multiplication tables (MSGT Ch. 1). b. Representations and character tables (MSGT Ch. 2). c. Irreducible representations and correlation tables (MSGT Ch. 3) d. Group theory approach to VB theory and hybrid orbitals (MSGT 4.1 to 4.2) 5.) Molecular orbital theory a. Linear combination of atomic orbitals, homonuclear diatomic molecules (IC 5.1 to 5.2.5) b. Polar and larger molecules (IC rest of Ch. 5). c. Symmetry adapted linear combination (SALC) orbitals (MSGT 4.3 to 4.5). -

Appendix 7 Tanabe-Sugano Diagrams and Some Illustrative Spectra
Appendix Consider the ligand ethylenediamine, en, which is typical of many. When it chelates to a metal atom a five-membered ring is formed, as shown in Fig. A 1.1. Is this ring planar? The answer is no, it is puckered. It is easy to see why this should be so. In a regular pentagon each internal angle is 108°, not far from the regular tetrahedron value of 109.SO. However, if the chelate ring is part of an octahedral complex then the N- M- N angle will be close to 900. This will open up the bond angles at the four atoms of the en ligand in the ring to an average of 112.5°; add to this the fact that the M- N bonds will be longer than the C--C and C- N and it is clear that some steric strain is likely to exist. This strain can be relieved at little or no cost if the ring puckers. Having recognized the possibility of puckering, the next thing is to recognize that two different puckered forms exist; much of this appendix is concerned with them. Any three points in space define a plane, unless the three points lie in a straight line. Consider the plane defined by the metal atom and the two nitrogen atoms of an en ligand in a complex. Our concern is with the two carbon atoms in the ligand. Relative to the plane, these can be arranged in either of the two ways shown in the top part of Figure A1.2. -

X Number of Four Centre M.H( Bonds. M
Pure & Appi. Chem.3 Vol.52, pp.7O5—7l2. 0033—4545/80/0301—0705$02.00/0 Pergamon Press Ltd. 1980. Printed in Great Britain. © IUPAC THEORETICALAND STRUCTURAL STUDIES ON ORGANOMETALLIC CLUSTER MOLECULES D. Michael P. Mingos Inorganic Chemistry Laboratory, South Parks Road, Oxford, OXI 3QR, England ABSTR,6CT. The development of topological rules for hydrido-metal cluster compounds, similar to those proposed by Lipscomb for boron hydrides, is shown to be a direct con- sequence of the multicentre bonding in bridged hydrido-metal bonds. Evidence for this localised view of the bonding has been obtained from U.V. photoelectron spectral studies of the hydrido- carbonyl cluster compounds Re3(CO)12H3, Os4(CO)12H4 and Os3(CO)10H2. The bonding in interstitial hydrido- cluster compounds, e.g. Co6(CO)15H, is also discussed in relation to the unusual chemi cal shift observed for the hydrido- ligand in this class of compound. Theoretical and preparative studies on metal centred cluster compounds, particularly of gold, are also discussed. A number of key structural determinations in the 1960's laid the foundations of our present understanding of the geometries of metal—hydrogen bonds in mononuclear transition metal complexes. X—ray investiga- tions (Refs. 1—3) showed hydrogen to be a stereochemi cally active liganci occupying a distinct coordina- tion site and two important neutron diffraction studies (Refs. 4 & 5) established that the M—H bond has a length of l.6-l.7A. More recently, X—ray and neutron studies on polynuclear metal cluster compounds with bridging hydrido- ligands particularly by Bau, Churchill, DahI, Williams and their coworkers (Ref.6) has resulted in a clearer definition of the geometric characteristics of the bridged metal—hydrogen bond and the suggestion that such bonds closely resemble the three centre two electron bond which has been established for The bridging hydrogen bonds in boranes (Ref. -

Ph.D. Syllabi 2019
COURSE SYLLABI FOR Ph.D. PROGRAM Course Syllabi (Ph.D.) Department of Chemistry Course Code: SCL604 of Oppenheimer approximation; vibrations of polyatomic molecules. Course Title: ROLE OF ORGANOMETALLIC COMPOUNDS IN Simple applications. ORGANIC SYNTHESIS Nuclear Magnetic Resonance: Nuclear Spin, nuclear resonance, Structure (L-T-P): 3-0-0-3 saturation, shielding of magnetic nuclei, chemical shift and its Pre-requisite: NIL measurements, factors influencing the chemical shift, Deshielding, spin- Contents: spin interaction, factors influencing coupling constant (J). Coupling – Organopalladium Chemistry: reactions involving organopalladium intermediates – palladium-catalyzed nucleophilic substitution and vicinal and geminal coupling, long-range coupling, spin decoupling, alkylation, Heck reaction, palladium-catalyzed cross coupling, and Spin systems (AX2, A2B2 & A2X2 and AMX, ABX, & ABC etc.), carbonylation reactions. NMR studies of nuclei other than proton: 13C, 19F and 31P. NOE, Organolithium reagents: Use of Organolithium in organic synthesis: n- simplification of complex spectra by the use of Shift reagent and field BuLi, s-BuLi, t-BuLi, Lithium di-isopropylamide (LDA) mediated strength. 2-D NMR, Introduction, NOESY, COSY, HETCOR. reactions. Mass Spectroscopy: Introduction, ion production, factors affecting Organocupper regents: Use of Organo Cupper in organic synthesis: fragmentation, ion analysis, ion abundance. Mass spectral fragmentation Gilman’s reagent, applications. Organoboron chemistry:carboranes, hydroboration, reaction of -

Inorganic Chemistry Ii
INORGANIC CHEMISTRY II OBJECTIVES 1. To understand the role of metal ions in biological process. 2. To learn the basic concepts of chemotherapy. 3. To learn the principle of catalysis and reaction mechanisms of organometallics. UNIT I: General Principles of Bioinorganic Chemistry Occurrence and availability of inorganic elements in biological systems – biomineralization – control and assembly of advanced materials in biology – nucleation and crystal growth – various biominerals – calcium phosphate – calcium carbonate – amorphous silica, iron biominerals – strontium and barium sulphate. Function and transport of alkali and alkaline earth metal ions: characterization of K+, Na+, Ca2+ and Mg2+ – complexes of alkali and alkaline earth metal ions with macrocycles – ion channels – ion pumps, catalysis and regulation of bioenergetic processes by the alkaline earth metal ions – Mg2+ and Ca2+. Metals at the center of photosynthesis – primary processes in photosynthesis – photosystems I and II-light absorption (energy acquisition) – exciton transport (direct energy transfer) – charge separation and electron transport – manganese catalyzed oxidation of water to O2. UNIT II: Amines, Proteins and Enzymes Cobalamines: reactions of the alkyl cobalamines – one electron reduction and oxidation – Co-C bond cleavage – coenzyme B12 – alkylation reactions of methylcobalamin. Heme and non-heme proteins – haemoglobin and myoglobin – oxygen transport and storage – electron transfer and oxygen activation – cytochromes, ferredoxins and rubredoxin – model systems, mononuclear non-heme iron enzymes. Copper containing proteins – classification and examples – electron transfer – oxygen transport-oxygenation – oxidases and reductases – cytochrome oxidase – superoxide dismutase (Cu, Zn) – nickel containing enzyme: urease. UNIT III: Medicinal Bioinorganic Chemistry Bioinorganic chemistry of quintessentially toxic metals – lead, cadmium, mercury, aluminium, chromium, copper and plutonium – detoxification by metal chelation – drugs that act by binding at the metal sites of metalloenzymes. -

Synthesis and Characterization of a Novel Triangular Rh2au Cluster
Full Papers ChemistryOpen doi.org/10.1002/open.202000217 1 2 3 Synthesis and Characterization of a Novel Triangular Rh2Au 4 5 Cluster Compound Inspired by the Isolobality Concept 6 [a] [a] [a] [b] 7 Hans-Christian Böttcher,* Marion Graf, Peter Mayer, and Manfred Scheer 8 Dedicated to Professor Paul Knochel on the occasion of his 65th birthday 9 10 5 5 11 The synthesis of [Rh2(η -Cp)2(μ-H)(μ-PPh2)2]BF4 (2) by protona- compound [Rh2{μ-Au(PPh3)}(η -Cp)2(μ-PPh2)2]BF4 (3) in good 5 12 tion reaction of the metal basic complex [Rh2(η -Cp)2(μ-PPh2)2] yield. Metal cluster salt 3 was fully characterized by spectro- 13 (1) with tetrafluoroboric acid in diethyl ether is described. scopic data and its molecular structure in the crystal was 14 Complex salt 2 was obtained in high yield and fully charac- determined by X-ray diffraction. The structural comparison of 15 terized by spectroscopic means and X-ray crystal diffraction. the protonated dirhodium core in the cationic complex of 2 16 Applying the isolobal analogy between H and the fragment Au with the Rh2Au framework in 3 is in good accordance with the + + 17 (PPh3) as a synthetic strategy on the reaction of compound 2 isolobal relation between H and Au because they share the 18 with equimolar amounts of [Au(CH3)(PPh3)] in refluxing acetone respective same position in these closely related molecular 19 resulted in the formation of the expected triangular cluster structures. 20 21 1. Introduction (PPh )}(η5-Cp)(CO) (μ-PPh )] could be realized using this path- 22 3 4 2 way. -

Generating Formulas of Transition Metal Carbonyl Clusters of Osmium, Rhodium and Rhenium
International Journal of Chemistry; Vol. 8, No. 1; 2016 ISSN 1916-9698 E-ISSN 1916-9701 Published by Canadian Center of Science and Education Generating Formulas of Transition Metal Carbonyl Clusters of Osmium, Rhodium and Rhenium Enos Masheija Rwantale Kiremire Correspondence: Enos Masheija Rwantale Kiremire, Department of Chemistry and Biochemistry, University of Namibia, Private Bag 13301, Windhoek, Namibia. E-mail:[email protected] Received: December 14, 2015 Accepted: December 30, 2015 Online Published: January 21, 2016 doi:10.5539/ijc.v8n1p126 URL: http://dx.doi.org/10.5539/ijc.v8n1p126 Abstract The series for carbonyl clusters of transition metals have been developed. They may be considered to be formed with a fragment centered around the 14 electron valence content. The capping series are based on the fragment of 12 electron valence content. The formulas of clusters can be decomposed into series from which the shapes of clusters may be predicted. The electron counting numbers of carbonyl clusters can be identified. Keywords: Carbonyl and borane clusters, fragments, isolobal principle, numerical sequence of fragments, building blocks, cluster series, 14n and 4n rules, valence electron content, cluster number 1. Introduction The discovery of the first mono-skeletal transition metal carbonyl Ni(CO)4 one hundred and twenty five years ago (Mond, et al, 1890), opened a wide gate for the synthesis of hundreds of metal carbonyl complexes. The structure of the nickel carbonyl Ni(CO)4 at the time was proposed to be as shown in Figure 1(F-1) in which the nickel atom exerted a valence of 2 while the valence of carbon and oxygen were 4 and 2 respectively. -

Electronic Structure of Clusters
Electronic Structure of Clusters David J. Wales Volume III, pp. 1506–1525 in Encyclopedia of Inorganic Chemistry, Second Edition (ISBN 0-470-86078-2) Editor-in-Chief : R. Bruce King John Wiley & Sons, Ltd, Chichester, 2005 material, but more than a single molecule or even two Electronic Structure of atoms or molecules’.2 Here, we must recognize that the domain of cluster chemistry extends beyond systems such Clusters as boranes, carboranes, and transition metal carbonyls, which are generally considered to be the stuff of inorganic cluster David J. Wales chemistry.1,3−5 These are certainly the molecules of primary University of Cambridge, Cambridge, UK interest here, but it would be shortsighted to ignore the connections between these species and clusters bound, for example, by van der Waals forces. There is often much to be learned from making such comparisons: for instance, we 1 Introduction 1 may find analogous structures and rearrangement mechanisms 2 Computational Approaches 1 in clusters bound by quite different forces. The converse is 3 Empirical Structure–Electron Count Correlations 2 also true, and a better understanding of the dynamics of small 4 Models of Cluster Bonding 3 inert gas clusters has been achieved partly by the application 5 Further Development of TSH Theory 8 of ideas that originated in inorganic chemistry concerning 6 Cluster Rearrangements 15 rearrangement mechanisms.6 7 Related Articles 18 To appreciate the ubiquity of clusters in chemistry and 8 References 18 physics, one need only consult the proceedings of one of the biennial International Symposia on Small Particles and Inorganic Clusters (ISSPIC).7 These volumes include species 8 Glossary ranging from hydrocarbon polyhedra, such as dodecahedrane (see also Dodecahedral), to large carbon clusters,9 and from small inert gas clusters to colloidal metal particles containing Ab initio calculations: attempt to calculate from first thousands of atoms. -

A Hypothetical Model for the Formation of Transition Metal Carbonyl Clusters Based Upon 4N Series Skeletal Numbers
International Journal of Chemistry; Vol. 8, No. 4; 2016 ISSN 1916-9698 E-ISSN 1916-9701 Published by Canadian Center of Science and Education A Hypothetical Model for the Formation of Transition Metal Carbonyl Clusters Based upon 4n Series Skeletal Numbers Enos Masheija Rwantale Kiremire Correspondence: Enos Masheija Rwantale Kiremire, Department of Chemistry and Biochemistry, University of Namibia, Private Bag 13301, Windhoek, Namibia. E-mail: [email protected] Received: August 23, 2016 Accepted: September 6, 2016 Online Published: September 29, 2016 doi:10.5539/ijc.v8n4p78 URL: http://dx.doi.org/10.5539/ijc.v8n4p78 Abstract Skeletal numbers of elements have been introduced as derivatives of the 4n series method. They are based on the number of valence electrons present in the skeletal element. They are extremely useful in deducing possible shapes of skeletal elements in molecules or clusters especially the small to medium ones. For large skeletal clusters, the skeletal numbers may simply be regarded as identity numbers. In carbonyl clusters, they can be used as a guide to facilitate the distribution of the ligands such as CO, H and charges onto the skeletal atoms. A naked skeletal cluster may be viewed as a reservoir for skeletal linkages which get utilized when ligands or electrons get bound to it. The sum of linkages used up by the ligands bound to a skeletal fragment and the remaining cluster skeletal numbers is equal to the number of the skeletal linkages present in the original „naked parent‟ skeletal cluster. The skeletal numbers can be used as a quick way of testing whether or not a skeletal atom obeys the 8-or 18-electron rules. -
From Intermetallics to Intermetalloid Clusters Molecular Alloying of Aluminum and Gallium with Transition Metals
Technische Universität München Lehrstuhl für Anorganische und Metallorganische Chemie From Intermetallics to Intermetalloid Clusters Molecular Alloying of Aluminum and Gallium with Transition Metals Jana Weßing Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Prof. Dr. Thomas F. Fässler Prüfende der Dissertation: 1. Prof. Dr. Roland A. Fischer 2. Prof. Dr. Jean-Yves Saillard 3. Prof. Dr. Stefanie Dehnen Die Dissertation wurde am 01.03.2018 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 30.04.2018 angenommen. FROM INTERMETALLICS TO INTERMETALLOID CLUSTERS Molecular Alloying of Aluminum and Gallium with Transition Metals Dissertation Jana Weßing Die vorliegende Arbeit wurde am Lehrstuhl für Anorganische Chemie II der Ruhr- Universität Bochum im Zeitraum von Januar 2014 bis März 2016, sowie am Lehrstuhl für Anorganische und Metallorganische Chemie der Technischen Universität München im Zeitraum von April 2016 bis März 2018 erstellt. Danksagung In erster Linie gilt mein besonderer Dank Prof. Dr. rer. nat. Dr. phil. h.c. Roland A. Fischer für die Möglichkeit mich in den letzten Jahren in Ihrer fachlich, wie menschlich herausragenden Arbeitsgruppe verwirklichen zu können. Ihnen verdanke ich nicht nur ein faszinierendes wissenschaftliches Thema, das meine Aufmerksamkeit trotz diverser Tiefschläge im Labor wecken und kontinuierlich fesseln konnte, sondern vor Allem die Gelegenheit alte Strukturen zu verlassen und mich in allen Belangen an neuen Herausforderungen messen und weiterentwickeln zu können. Für Ihr mir dabei entgegengebrachtes Vertrauen und Ihre Unterstützung bin ich von Herzen dankbar. Meiner Prüfungskommission danke ich für die bereitwillige und freundliche Übernahme des Koreferats. -
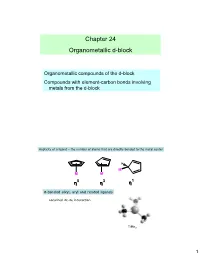
Chapter 24 Organometallic D-Block
Chapter 24 Organometallic d-block Organometallic compounds of the d-block Compounds with element-carbon bonds involving metals from the d-block Hapticity of a ligand – the number of atoms that are directly bonded to the metal center H M M M ηηη5 ηηη3 ηηη1 σ-bonded alkyl, aryl and related ligands Localized 2c-2e interaction TiMe 3 1 Dewar-Chatt-Duncanson model 2 semi-bridging In multinuclear metal species a number of bonding modes may be adopted. + : M − C ≡ O : ↔ M = C = O :: υ -1 Free CO, CO 2143 cm d(CO) = 112.8 pm υ -1 M-C (cm ) 416 441 3 Hydride ligands 3c-2e 4c-2e 7c-2e interstitial 4 Metal complexes with H 2 Monodentate organophosphines: σ-donor and π-acceptor tertiary: PR 3 secondary: PR 2H primary: PRH 2 π-accepting properties: t PF 3 > P(OPh) 3 > P(OMe) 3 > PPh 3 > P Bu 3 5 π-bonded ligands 6 146 pm 134 pm 134 pm 138 pm 143 pm 141 pm 3 4 5 free buta-1,3-diene Mo(η -C3H5)(η -C4H6)(η -C5H5) 7 Nitrogen monoxide Dinitrogen •radical •N2 and CO are •singly bound as nitrosyl ligand isoelectronic, similar •linear or bent (165-180°) bonding •donates three electrons to metal •Complexes of N 2 not as υ -1 stable as CO • NO 1525-1690 cm M=N=O :M-NΞO: Dihydrogen •σ-MO (electron donor O orbital) and σ*-MO M-N (acceptor) •can weaken or cleave the H-H bond 8 18-electron rule •Low oxidation state organometallic complexes tend to obey the 18- electron rule. -

Electron Rule. Q the Formal Description of Nitric Oxide As NO+ Does Not Match Certain Measureable and Calculated Properties
INORGANIC CHEMISTRY-I Dr. V.RAJASUDHA. M.Sc., M.Phil., B.Ed. Ph.D., 2 Scope of the present work 18 -electron rule 1 Isolopal concept and its usefulness Nitroysyl complxes Metallocene and arene complexes Application of organometallics Y D U To unterstand the role of metal ions in biological process T S E H T F To learn the basic concept of alkyl metals. O S E V I To learn the principle of cataiysis and reaction T C mechanism of organomatallics. E J B O 4 18-Electron Rule v The 18-electron rule is a rule used primarily for predicting and rationalizing formulas for stable metal complexes, especially organometallic compounds. v The rule is based on the fact that the valence shells of transition metals consist of nine valence orbitals (one s orbital, three p orbitals and five d orbitals), which collectively can accommodate 18 electrons as either bonding or nonbonding electron pairs. v This means that the combination of these nine atomic orbitals with ligand orbitals creates nine molecular orbitals that are either metal-ligand bonding or non-bonding. v When a metal complex has 18 valence electrons, it is said to have achieved the same electron configuration as the noble gas in the period. v The rule and its exceptions are similar to the application of the octet rule to main group elements. v The rule is not helpful for complexes of metals that are not transition metals, and interesting or useful transition metal complexes will violate the rule because of the consequences deviating from the rule bears on reactivity.