And IRAK1 Knock-In Mice Production Defined by the Study of IRAK2 Two
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carna Newsletter 2021.05.26
画像:ファイル>配置>リンクから画像を選択>画像を選択して右クリック>画像を最前面に配置>オブジェクト>テキストの回り込み>作成 Vol.10 Carna Newsletter 2021.05.26 Targeted Degradation of Non-catalytic Kinases New Drug Discovery Options The announcement that Kymera Therapeutics, a TLR/IL-1R signaling in several cell types, which company pioneering targeted protein degradation, indicates that the IRAK4 scaffolding function is entered into a strategic collaboration with Sanofi important in some cases2)3). to develop and commercialize first-in-class protein degrader therapies targeting IRAK4 in patients with immune-inflammatory diseases highlights the TLR growing interest in clinical applications of small IL-1R molecule mediated kinase degradation. Kymera received $150 million in cash up front and may cytosol 8 8 potentially receive at least $2 billion in this key D y strategic partnership, including sales milestones M 4 4 K K and royalty payments. A A R R I I 2 P 1 K Most of the protein degraders currently under K A A R I R development are heterobifunctional molecules I which contain one moiety that binds a desired target protein and another that binds an E3 ligase, P joined by a linker. Protein degrader-induced proximity results in ubiquitination of the target Fig.1. TLR and IL-1R Signaling in the Myddosome followed by its degradation by the proteasome. This transformative new modality is expected to open a new chapter in drug discovery targeting 【Allosteric regulatory function】 kinases for which the development of clinical Aside from scaffolding functions, allosteric inhibitors has been difficult. One such example is regulation is another non-catalytic function that IRAK4, which has a non-catalytic function activates binding partners by inducing a independent of kinase activity in addition to a conformational change. -

Recombinant Human IFN-Α6 Mammalian Cell-Expressed Human Interferon Alpha 6 (Alpha K) with HSA Cat
Recombinant human IFN-α6 Mammalian cell-expressed human interferon alpha 6 (alpha K) with HSA Cat. code: rcyc-hifna6 https://www.invivogen.com/human-ifna6 For research use only, not for diagnostic or therapeutic use Version 18I12-NJ PRODUCT INFORMATION BACKGROUND Contents Type I interferons (IFN) include the IFN-α family, IFN-β, IFN-ε, IFN-κ • 1 µg of lyophilized recombinant human IFN-α6 and IFN-ω. IFN-αs are important anti-viral cytokines that also play Note: This product is sterile filtered prior to lyophilization. anti-proliferative and immuno-modulatory functions. The human IFN-α • 1.5 ml endotoxin-free water family comprises 13 genes encoding 12 proteins, IFN-α13 being identical to IFN-α1. All IFN-αs bind to a common heterodimer receptor IFNAR1/ Storage and stability IFNAR2. The ternary complex signals through the Janus kinase (JAK) • Recombinant human IFN-α6 is shipped at room temperature. Upon and signal transducer and activator of transcription (STAT) signaling receipt it should be immediately stored at -20 °C. Lyophilized product is stable for 6 months at -20°C. pathway, inducing the formation of the ISGF3 transcriptional complex • Upon resuspension, prepare aliquots of recombinant human IFN-α6 and (STAT1/STAT2/IRF9). ISGF3 binds to IFN-stimulated response elements 2 store at -80 °C for 4 months. (ISRE) in the promoters of numerous IFN-stimulated genes (ISGs) . Note: Avoid repeated freeze-thaw cycles. Quality control IFN-α IFN-α • Purity: ≥ 95% (SDS-PAGE) Low affinity High affinity • Endotoxin: ≤ 1 EU/µg • The biological activity has been confirmed using HEK-Blue™ IFN-α/β IFNAR1 IFNAR2 cells (see validation data sheet available on our website). -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Prevalent Homozygous Deletions of Type I Interferon and Defensin Genes in Human Cancers Associate with Immunotherapy Resistance
Author Manuscript Published OnlineFirst on April 4, 2018; DOI: 10.1158/1078-0432.CCR-17-3008 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Prevalent Homozygous Deletions of Type I Interferon and Defensin Genes in Human Cancers Associate with Immunotherapy Resistance Zhenqing Ye1; Haidong Dong2,3; Ying Li1; Tao Ma1,4; Haojie Huang4; Hon-Sing Leong4; Jeanette Eckel-Passow1; Jean-Pierre A. Kocher1; Han Liang5; LiguoWang1,4,* 1Division of Biomedical Statistics and Informatics, Department of Health Sciences, Mayo Clinic, Rochester, Minnesota 55905, United States of America 2Department of Immunology, College of Medicine, Mayo Clinic, Rochester, Minnesota 55905, United States of America 3Department of Urology, Mayo Clinic, Rochester, Minnesota 55905, United States of America 4Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, Minnesota 55905, United States of America 5Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, United States of America Running title: Pervasive Deletion of Interferon/Defensin in Human Cancers Keywords: Homozygous deletion, Type-I interferon, Defensin, immunotherapy resistance, cancer * Correspondence to: Liguo Wang, Ph.D. Associate Professor Division of Biomedical Statistics and Informatics, Mayo Clinic 200 1st St SW Rochester, MN 55905, USA Phone: +1-507-284-8728 Fax: +1-507-284-0745 Email: [email protected] The authors declare no potential conflicts of interest. 1 Downloaded from clincancerres.aacrjournals.org on September 29, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on April 4, 2018; DOI: 10.1158/1078-0432.CCR-17-3008 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

( 12 ) United States Patent
US010428349B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 , 428 ,349 B2 DeRosa et al . (45 ) Date of Patent: Oct . 1 , 2019 ( 54 ) MULTIMERIC CODING NUCLEIC ACID C12N 2830 / 50 ; C12N 9 / 1018 ; A61K AND USES THEREOF 38 / 1816 ; A61K 38 /45 ; A61K 38/ 44 ; ( 71 ) Applicant: Translate Bio , Inc ., Lexington , MA A61K 38 / 177 ; A61K 48 /005 (US ) See application file for complete search history . (72 ) Inventors : Frank DeRosa , Lexington , MA (US ) ; Michael Heartlein , Lexington , MA (56 ) References Cited (US ) ; Daniel Crawford , Lexington , U . S . PATENT DOCUMENTS MA (US ) ; Shrirang Karve , Lexington , 5 , 705 , 385 A 1 / 1998 Bally et al. MA (US ) 5 ,976 , 567 A 11/ 1999 Wheeler ( 73 ) Assignee : Translate Bio , Inc ., Lexington , MA 5 , 981, 501 A 11/ 1999 Wheeler et al. 6 ,489 ,464 B1 12 /2002 Agrawal et al. (US ) 6 ,534 ,484 B13 / 2003 Wheeler et al. ( * ) Notice : Subject to any disclaimer , the term of this 6 , 815 ,432 B2 11/ 2004 Wheeler et al. patent is extended or adjusted under 35 7 , 422 , 902 B1 9 /2008 Wheeler et al . 7 , 745 ,651 B2 6 / 2010 Heyes et al . U . S . C . 154 ( b ) by 0 days. 7 , 799 , 565 B2 9 / 2010 MacLachlan et al. (21 ) Appl. No. : 16 / 280, 772 7 , 803 , 397 B2 9 / 2010 Heyes et al . 7 , 901, 708 B2 3 / 2011 MacLachlan et al. ( 22 ) Filed : Feb . 20 , 2019 8 , 101 ,741 B2 1 / 2012 MacLachlan et al . 8 , 188 , 263 B2 5 /2012 MacLachlan et al . (65 ) Prior Publication Data 8 , 236 , 943 B2 8 /2012 Lee et al . -
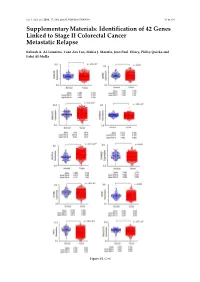
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

IRAK2, Active IRAK2, Active
Catalogue # Aliquot Size I10-10BG-05 5 µg I10-10BG-10 10 µg I10-10BG-20 20 µg IRAK2, Active Full-length recombinant protein expressed in Sf9 cells Catalog # I10-10BG Lot # V117 -2B Product Description Specific Activity Recombinant full-length human IRAK2 was expressed by 280,000 baculovirus in Sf9 insect cells using an N-terminal GST tag. The gene accession number is NM_001570 . 210,000 Gene Aliases 140,000 IRAK-2, MGC150550 70,000 (RLU) Activity Formulation 0 0 200 400 600 800 Recombinant protein stored in 50mM Tris-HCl, pH 7.5, Protein (ng) 150mM NaCl, 10mM glutathione, 0.1mM EDTA, 0.25mM DTT, 0.1mM PMSF, 25% glycerol. The specific activity of IRAK2 was determined to be 6 nmol /min/mg as per activity assay protocol. Storage and Stability Purity o Store product at –70 C. For optimal storage, aliquot target into smaller quantities after centrifugation and store at recommended temperature. For most favorable performance, avoid repeated handling and multiple freeze/thaw cycles. The purity of IRAK2 was determined to be >75% by densitometry, approx. MW 103kDa . Scientific Background Interleukin-1 receptor-associated kinase 2 (IRAK2) is important downstream signaling components of Toll-like receptors (TLRs) (1). IRAKs were first described as signal transducers for IL-1 and later have been implicated in signal transduction of other members of the Toll/IL-1 IRAK2, Active receptor family. The interleukin-1 receptor (IL-1R) signaling Full-length recombinant protein expressed in Sf9 cells pathway leads to NF κB activation in mammals. To date, Catalog Number I10-10BG four mammalian IRAKs have been identified (IRAK-1, Specific Activity 6 nmol/min/mg IRAK-2, IRAK-4, and IRAK-M) (2). -

Single Cell Derived Clonal Analysis of Human Glioblastoma Links
SUPPLEMENTARY INFORMATION: Single cell derived clonal analysis of human glioblastoma links functional and genomic heterogeneity ! Mona Meyer*, Jüri Reimand*, Xiaoyang Lan, Renee Head, Xueming Zhu, Michelle Kushida, Jane Bayani, Jessica C. Pressey, Anath Lionel, Ian D. Clarke, Michael Cusimano, Jeremy Squire, Stephen Scherer, Mark Bernstein, Melanie A. Woodin, Gary D. Bader**, and Peter B. Dirks**! ! * These authors contributed equally to this work.! ** Correspondence: [email protected] or [email protected]! ! Supplementary information - Meyer, Reimand et al. Supplementary methods" 4" Patient samples and fluorescence activated cell sorting (FACS)! 4! Differentiation! 4! Immunocytochemistry and EdU Imaging! 4! Proliferation! 5! Western blotting ! 5! Temozolomide treatment! 5! NCI drug library screen! 6! Orthotopic injections! 6! Immunohistochemistry on tumor sections! 6! Promoter methylation of MGMT! 6! Fluorescence in situ Hybridization (FISH)! 7! SNP6 microarray analysis and genome segmentation! 7! Calling copy number alterations! 8! Mapping altered genome segments to genes! 8! Recurrently altered genes with clonal variability! 9! Global analyses of copy number alterations! 9! Phylogenetic analysis of copy number alterations! 10! Microarray analysis! 10! Gene expression differences of TMZ resistant and sensitive clones of GBM-482! 10! Reverse transcription-PCR analyses! 11! Tumor subtype analysis of TMZ-sensitive and resistant clones! 11! Pathway analysis of gene expression in the TMZ-sensitive clone of GBM-482! 11! Supplementary figures and tables" 13" "2 Supplementary information - Meyer, Reimand et al. Table S1: Individual clones from all patient tumors are tumorigenic. ! 14! Fig. S1: clonal tumorigenicity.! 15! Fig. S2: clonal heterogeneity of EGFR and PTEN expression.! 20! Fig. S3: clonal heterogeneity of proliferation.! 21! Fig. -

Specialized Interferon Ligand Action in COVID19
medRxiv preprint doi: https://doi.org/10.1101/2021.07.29.21261325; this version posted August 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . Specialized interferon ligand action in COVID19 Matthew D. Galbraith1,2, Kohl T. Kinning1, Kelly D. Sullivan1,3, Paula Araya1, Keith P. Smith1, Ross E. Granrath1, Jessica R. Shaw1, Ryan Baxter4, Kimberly R. Jordan4, Seth Russell5, Monika Dzieciatkowska6, Julie A. Reisz6, Fabia Gamboni6, Francesca CenDali6, Tusharkanti Ghosh7, AnDrew A. Monte8, Tellen D. Bennett9, Kirk C. Hansen6, Elena W.Y. Hsieh4,10, Angelo D’AlessanDro6, and Joaquin M. Espinosa1,2* Affiliations: 1Linda Crnic Institute for Down SynDrome, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 2Department of Pharmacology, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 3Department of PeDiatrics, Section of Developmental Biology, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 4Department of Immunology anD Microbiology, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 5Data Science to Patient Value, University of Colorado Anschutz Medical Campus; Aurora, CO, USA. 6Department of Biochemistry anD Molecular Genetics, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 7Department of Biostatistics anD Informatics, ColoraDo School of Public Health; Aurora, CO, USA. 8Department of Emergency MeDicine, University of ColoraDo Anschutz MeDical Campus; Aurora, CO, USA. 9Department of PeDiatrics, Sections of Informatics anD Data Science anD Critical Care MeDicine, University of ColoraDo Anschutz Medical Campus; Aurora, CO, USA. -

IRAK2 Directs Stimulus-Dependent Nuclear Export of Inflammatory Mrnas
IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Zhou, H., K. Bulek, X. Li, T. Herjan, M. Yu, W. Qian, H. Wang, et al. 2017. “IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs.” eLife 6 (1): e29630. doi:10.7554/eLife.29630. http://dx.doi.org/10.7554/eLife.29630. Published Version doi:10.7554/eLife.29630 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:34493075 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA RESEARCH ARTICLE IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs Hao Zhou1†, Katarzyna Bulek1,2†, Xiao Li3†, Tomasz Herjan1†, Minjia Yu1,4, Wen Qian1, Han Wang1, Gao Zhou3, Xing Chen1, Hui Yang1, Lingzi Hong1, Junjie Zhao1, Luke Qin1, Koichi Fukuda5, Annette Flotho6, Ji Gao7, Ashok Dongre7, Julie A Carman7, Zizhen Kang1,8,9, Bing Su8,9,10, Timothy S Kern11,12, Jonathan D Smith13, Thomas A Hamilton1, Frauke Melchior6, Paul L Fox13, Xiaoxia Li1* 1Department of Immunology, Lerner Research Institute, Cleveland Clinic, Cleveland, United States; 2Department of Immunology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Krakow, Poland; 3Department of Genetics, Stanford University School of Medicine, -

Interferonsource.Com What's Inside New Products Research Tools Protocols & Tips Product Citations What's Your IFN-Α
Interferon Matters A quarterly newsletter by PBL InterferonSource interferonsource.com May 2007, Issue 2 u What’s Inside What’s Your IFN-a Type? New Products Are all Type I IFNs created equal? Of Mice and Men page 4 & 5 Type I Interferons were the first cytokines to be The biological significance of the expression of discovered. They are a family of homologous cytokines so many different IFN-a subtypes has yet to be Rhesus/Cynomolgus originally identified for their antiviral activity but have determined. However, reports suggest that they show IFN-a2 Subtype also been shown to have both anti-proliferative and quantitatively distinct anti-viral, anti-proliferative, immunomodulatory activities. In humans and mice and killer cell-stimulatory activities. It is therefore Mouse IFN-a4 Subtype they are encoded by an intronless multigene family of importance for researchers to characterize and clustered on human chromosome 9 and murine study these IFN-a subtypes and their possible post- chromosome 4 respectively. translational modifications. Research Tools Also in both species there are multiple Interferon- The common models for IFN-a subtypes study are page 6 & 7 Alpha (IFN-a) genes and one Interferon-Beta (IFN-b) human and mouse. In 1998, Lawson et al showed that Human IFN-a gene. Other Type I Interferon genes described include there are in vivo differences in the antiviral activity of the Interferon-Omega (IFN-w) gene, murine limitin the mouse IFN-a subtypes.1 Using an experimental Multi-Subtype ELISA Kit gene, as well as the human/murine Interferon-Epilson, mouse model of IFN transgene expression in vivo, TM Interferon-Kappa, and Interferon-Tau (IFN-e, IFN-k, and the authors showed that mouse IFN-a1 has a greater iLite Human IFN-a Kit IFN-t).