Comprehensive in Vivo Identification of the C-Myc Mrna Interactome Using Hypr-MS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Topological Scoring of Protein Interaction Networks
bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Topological Scoring of Protein Interaction Networks Mihaela E. Sardiu1, Joshua M. Gilmore1,2, Brad D. Groppe1,3, Arnob Dutta1,4, Laurence Florens1, and Michael P. Washburn1,5‡ 1Stowers Institute for Medical Research, Kansas City, MO 64110 U.S.A. 2Current Address: Boehringer Ingelheim Vetmedica, St. Joseph, MO 64506 U.S.A. 3Current Address: Thermo Fisher Scientific, Waltham, MA 02451, U.S.A. 4Current Address: Department of Cell and Molecular Biology, University of Rhode Island, 287 CBLS, 120 Flagg Road, Kingston, RI 02881. 5 Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, Kansas 66160, USA ‡To whom correspondence should be addressed: Michael Washburn, Ph.D. Stowers Institute for Medical Research 1000 E. 50th St. Kansas City, MO 64110 Phone: 816-926-4457 E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract It remains a significant challenge to define individual protein associations within networks where an individual protein can directly interact with other proteins and/or be part of large complexes, which contain functional modules. Here we demonstrate the topological scoring (TopS) algorithm for the analysis of quantitative proteomic analyses of affinity purifications. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Revealing the Mechanism of Xist-Mediated Silencing
Revealing the Mechanism of Xist-mediated Silencing Thesis by Chun-Kan Chen In Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2018 Defended November 1, 2017 ii 2017 Chun-Kan Chen ORCID: 0000-0002-1194-9137 iii ACKNOWLEDGEMENTS First of all, I’d like to thank my great mentor, Dr. Mitch Guttman (California Institute of Technology, Pasadena, CA), who led me to become an independent researcher and gave me valuable advice that guided me to accomplish this thesis. He has always been supportive of my future plans and career goals. I really enjoyed every discussion we have had. We often generated some interesting ideas for projects during our discussions. I would also like to send my thanks to my lab mates, Amy Chow, Mario Blanco, and Erik Aznauryan, who helped me with many experiments to move the project forward. I’d like to acknowledge Dr. Kathrin Plath (University of California, Los Angeles, Los Angeles, CA) for the collaboration and his critical comments on this project. Also, I want to thank Jesse Engreitz and Patrick McDonel, who provided helpful comments and suggestions to the project. I want to thank my parents, brother, and parents-in-law who provided both instrumental and emotional support to assist me in completing my Ph.D. degree. I also want to thank my friends, Lily Chen, Pei-Ying Lin, Tzu-Yao Wang, and Wei Li, for giving me valuable social support during my years in graduate school. Last but not least, I would like to send my special thanks to my wife, Christine Juang, who has always been supportive. -

Urabe VK, Et Al. Influences on U2 Snrna Structure U2
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.05.451154; this version posted July 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Urabe VK, et al. Influences on U2 snRNA structure U2 snRNA structure is influenced by SF3A and SF3B proteins but not by SF3B inhibitors Veronica K. Urabe1, Meredith Stevers1, Arun K. Ghosh3 and Melissa S. Jurica1,2 * 1Department of Molecular Cell and Developmental Biology and 2Center for Molecular Biology of RNA, University of California, Santa Cruz, California, United States of America 3Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana United States of America *Corresponding author E-mail: [email protected] (MSJ) bioRxiv preprint doi: https://doi.org/10.1101/2021.07.05.451154; this version posted July 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Urabe VK, et al. Influences on U2 snRNA structure Abstract U2 snRNP is an essential component of the spliceosome. It is responsible for branch point recognition in the spliceosome A-complex via base-pairing of U2 snRNA with an intron to form the branch helix. Small molecule inhibitors target the SF3B component of the U2 snRNP and interfere with A-complex formation during spliceosome assembly. -

SF3B2 Monoclonal Antibody (M01J), Clone 5D2
SF3B2 monoclonal antibody (M01J), clone 5D2 Catalog # : H00010992-M01J 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Cell lysate) Description: SF3B2. Immunogen: SF3B2 (NP_006833, 592 a.a. ~ 645 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: YEGKEFETRLKEKKPGDLSDELRISLGMPVGPNAHKVPPPWLIAMQRYG PPPSY enlarge Western Blot (Cell lysate) Host: Mouse Reactivity: Human, Mouse, Rat Preparation Cell Culture Production Method: (CX Grade Antibody List) enlarge Isotype: IgG2a Kappa Western Blot (Recombinant protein) Quality Control Antibody Reactive Against Recombinant Protein. Immunofluorescence Testing: enlarge Immunohistochemistry (Formalin/PFA-fixed paraffin- embedded sections) Western Blot detection against Immunogen (31.68 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. enlarge Instruction: Sandwich ELISA (Recombinant protein) MSDS: Download Interspecies Mouse (98); Rat (98) Antigen Sequence: Datasheet: Download enlarge ELISA Applications Western Blot (Cell lysate) Page 1 of 3 2021/6/15 SF3B2 monoclonal antibody (M01J), clone 5D2. Western Blot analysis of SF3B2 expression in Hela S3 NE. Protocol Download Western Blot (Cell lysate) SF3B2 monoclonal antibody (M01J), clone 5D2. Western Blot analysis of SF3B2 expression in Jurkat. Protocol Download Western Blot (Recombinant protein) Protocol Download Immunofluorescence enlarge this image Immunofluorescence of monoclonal antibody to SF3B2 on HeLa cell . [antibody concentration 10 ug/ml] Immunohistochemistry (Formalin/PFA-fixed paraffin-embedded sections) enlarge this image Page 2 of 3 2021/6/15 Immunoperoxidase of monoclonal antibody to SF3B2 on formalin-fixed paraffin-embedded human kidney. [antibody concentration 6 ug/ml] Protocol Download Sandwich ELISA (Recombinant protein) Detection limit for recombinant GST tagged SF3B2 is 0.1 ng/ml as a capture antibody. -

Analysis of the Stability of 70 Housekeeping Genes During Ips Reprogramming Yulia Panina1,2*, Arno Germond1 & Tomonobu M
www.nature.com/scientificreports OPEN Analysis of the stability of 70 housekeeping genes during iPS reprogramming Yulia Panina1,2*, Arno Germond1 & Tomonobu M. Watanabe1 Studies on induced pluripotent stem (iPS) cells highly rely on the investigation of their gene expression which requires normalization by housekeeping genes. Whether the housekeeping genes are stable during the iPS reprogramming, a transition of cell state known to be associated with profound changes, has been overlooked. In this study we analyzed the expression patterns of the most comprehensive list to date of housekeeping genes during iPS reprogramming of a mouse neural stem cell line N31. Our results show that housekeeping genes’ expression fuctuates signifcantly during the iPS reprogramming. Clustering analysis shows that ribosomal genes’ expression is rising, while the expression of cell-specifc genes, such as vimentin (Vim) or elastin (Eln), is decreasing. To ensure the robustness of the obtained data, we performed a correlative analysis of the genes. Overall, all 70 genes analyzed changed the expression more than two-fold during the reprogramming. The scale of this analysis, that takes into account 70 previously known and newly suggested genes, allowed us to choose the most stable of all genes. We highlight the fact of fuctuation of housekeeping genes during iPS reprogramming, and propose that, to ensure robustness of qPCR experiments in iPS cells, housekeeping genes should be used together in combination, and with a prior testing in a specifc line used in each study. We suggest that the longest splice variants of Rpl13a, Rplp1 and Rps18 can be used as a starting point for such initial testing as the most stable candidates. -

HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins
HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins This information is current as Adi Reches, Daphna Nachmani, Orit Berhani, Alexandra of September 29, 2021. Duev-Cohen, Dorin Shreibman, Yael Ophir, Barbara Seliger and Ofer Mandelboim J Immunol 2016; 196:4967-4976; Prepublished online 18 May 2016; doi: 10.4049/jimmunol.1501550 Downloaded from http://www.jimmunol.org/content/196/12/4967 References This article cites 34 articles, 6 of which you can access for free at: http://www.jimmunol.org/content/196/12/4967.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 29, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins Adi Reches,* Daphna Nachmani,* Orit Berhani,* Alexandra Duev-Cohen,* Dorin Shreibman,* Yael Ophir,* Barbara Seliger,† and Ofer Mandelboim* MHC class I molecules, in addition to their role in specific activation of the CTL of adaptive immune system, function also as the main ligands for NK cell inhibitory receptors, which prevent NK cells from killing normal, healthy cells. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Upon Microbial Challenge, Human Neutrophils Undergo Rapid Changes in Nuclear Architecture and Chromatin Folding to Orchestrate an Immediate Inflammatory Gene Program
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Upon microbial challenge, human neutrophils undergo rapid changes in nuclear architecture and chromatin folding to orchestrate an immediate inflammatory gene program Matthew Denholtz,1,5 Yina Zhu,1,5 Zhaoren He,1 Hanbin Lu,1 Takeshi Isoda,1,4 Simon Döhrmann,2 Victor Nizet,2,3 and Cornelis Murre1 1Division of Biological Sciences, Department of Molecular Biology, University of California at San Diego, La Jolla, California 92039, USA; 2Department of Pediatrics, University of California at San Diego School of Medicine, La Jolla, California 92093, USA; 3Skaggs School of Pharmaceutical Sciences, University of California at San Diego, La Jolla, California 92093, USA Differentiating neutrophils undergo large-scale changes in nuclear morphology. How such alterations in structure are established and modulated upon exposure to microbial agents is largely unknown. Here, we found that prior to encounter with bacteria, an armamentarium of inflammatory genes was positioned in a transcriptionally passive environment suppressing premature transcriptional activation. Upon microbial exposure, however, human neu- trophils rapidly (<3 h) repositioned the ensemble of proinflammatory genes toward the transcriptionally permissive compartment. We show that the repositioning of genes was closely associated with the swift recruitment of cohesin across the inflammatory enhancer landscape, permitting an immediate transcriptional response upon bacterial exposure. We found that activated enhancers, marked by increased deposition of H3K27Ac, were highly enriched for cistromic elements associated with PU.1, CEBPB, TFE3, JUN, and FOSL2 occupancy. These data reveal how upon microbial challenge the cohesin machinery is recruited to an activated enhancer repertoire to instruct changes in chromatin folding, nuclear architecture, and to activate an inflammatory gene program. -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g. -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -
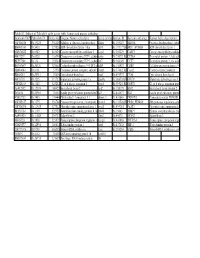
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1