Universidade De Lisboa Faculdade De Ciências
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FUSIP1 Polyclonal Antibody Catalog Number PA5-41929 Product Data Sheet
Lot Number: A9C701N Website: thermofisher.com Customer Service (US): 1 800 955 6288 ext. 1 Technical Support (US): 1 800 955 6288 ext. 441 thermofisher.com/contactus FUSIP1 Polyclonal Antibody Catalog Number PA5-41929 Product Data Sheet Details Species Reactivity Size 100 µl Tested species reactivity Equine, Guinea Pig, Human, Mouse, Host / Isotype Rabbit IgG Rabbit, Rat Class Polyclonal Tested Applications Dilution * Type Antibody Immunohistochemistry (Paraffin) 4-8 µg/mL (IHC (P)) Immunogen Synthetic peptide directed towards the C-terminal of human FUSIP1 Western Blot (WB) 0.2-1 µg/mL Conjugate Unconjugated * Suggested working dilutions are given as a guide only. It is recommended that the user titrate the product for use in their own experiment using appropriate negative and positive controls. Form Liquid Concentration 0.5mg/mL Purification Affinity Chromatography Storage Buffer PBS with 2% sucrose Contains 0.09% sodium azide Storage Conditions -20° C, Avoid Freeze/Thaw Cycles Product Specific Information Peptide sequence: TDSKTHYKSG SRYEKESRKK EPPRSKSQSR SQSRSRSKSR SRSWTSPKSS Sequence homology: Guinea Pig: 100%; Horse: 100%; Human: 100%; Mouse: 100%; Rabbit: 100%; Rat: 100% Background/Target Information FUSIP1 is a member of the serine-arginine (SR) family of proteins, which is involved in constitutive and regulated RNA splicing. Members of this family are characterized by N-terminal RNP1 and RNP2 motifs, which are required for binding to RNA, and multiple C-terminal SR/RS repeats, which are important in mediating association with other cellular proteins. This protein can influence splice site selection of adenovirus E1A pre-mRNA. It interacts with the oncoprotein TLS, and abrogates the influence of TLS on E1A pre-mRNA splicing.This gene product is a member of the serine-arginine (SR) family of proteins, which is involved in constitutive and regulated RNA splicing. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

FUSIP1 (SRSF10) (NM 006625) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC221759 FUSIP1 (SRSF10) (NM_006625) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: FUSIP1 (SRSF10) (NM_006625) Human Tagged ORF Clone Tag: Myc-DDK Symbol: SRSF10 Synonyms: FUSIP1; FUSIP2; NSSR; PPP1R149; SFRS13; SFRS13A; SRp38; SRrp40; TASR; TASR1; TASR2 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC221759 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGTCCCGCTACCTGCGTCCCCCCAACACGTCTCTGTTCGTCAGGAACGTGGCCGACGACACCAGGTCTG AAGACTTGCGGCGTGAATTTGGTCGTTATGGTCCTATAGTTGATGTGTATGTTCCACTTGATTTCTACAC TCGCCGTCCAAGAGGATTTGCTTATGTTCAATTTGAGGATGTTCGTGATGCTGAAGACGCTTTACATAAT TTGGACAGAAAGTGGATTTGTGGACGGCAGATTGAAATACAGTTTGCCCAGGGGGATCGAAAGACACCAA ATCAGATGAAAGCCAAGGAAGGGAGGAATGTGTACAGTTCTTCACGCTATGATGATTATGACAGATACAG ACGTTCTAGAAGCCGAAGTTATGAAAGGAGGAGATCAAGAAGTCGGTCTTTTGATTACAACTATAGAAGA TCGTATAGTCCTAGAAACAGTAGACCGACTGGAAGACCACGGCGTAGCAGAAGCCATTCCGACAATGATA GACCAAACTGCAGCTGGAATACCCAGTACAGTTCTGCTTACTACACTTCAAGAAAGATC ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA Protein Sequence: >RC221759 protein sequence Red=Cloning site Green=Tags(s) MSRYLRPPNTSLFVRNVADDTRSEDLRREFGRYGPIVDVYVPLDFYTRRPRGFAYVQFEDVRDAEDALHN -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Location Analysis of Estrogen Receptor Target Promoters Reveals That
Location analysis of estrogen receptor ␣ target promoters reveals that FOXA1 defines a domain of the estrogen response Jose´ e Laganie` re*†, Genevie` ve Deblois*, Ce´ line Lefebvre*, Alain R. Bataille‡, Franc¸ois Robert‡, and Vincent Gigue` re*†§ *Molecular Oncology Group, Departments of Medicine and Oncology, McGill University Health Centre, Montreal, QC, Canada H3A 1A1; †Department of Biochemistry, McGill University, Montreal, QC, Canada H3G 1Y6; and ‡Laboratory of Chromatin and Genomic Expression, Institut de Recherches Cliniques de Montre´al, Montreal, QC, Canada H2W 1R7 Communicated by Ronald M. Evans, The Salk Institute for Biological Studies, La Jolla, CA, July 1, 2005 (received for review June 3, 2005) Nuclear receptors can activate diverse biological pathways within general absence of large scale functional data linking these putative a target cell in response to their cognate ligands, but how this binding sites with gene expression in specific cell types. compartmentalization is achieved at the level of gene regulation is Recently, chromatin immunoprecipitation (ChIP) has been used poorly understood. We used a genome-wide analysis of promoter in combination with promoter or genomic DNA microarrays to occupancy by the estrogen receptor ␣ (ER␣) in MCF-7 cells to identify loci recognized by transcription factors in a genome-wide investigate the molecular mechanisms underlying the action of manner in mammalian cells (20–24). This technology, termed 17-estradiol (E2) in controlling the growth of breast cancer cells. ChIP-on-chip or location analysis, can therefore be used to deter- We identified 153 promoters bound by ER␣ in the presence of E2. mine the global gene expression program that characterize the Motif-finding algorithms demonstrated that the estrogen re- action of a nuclear receptor in response to its natural ligand. -
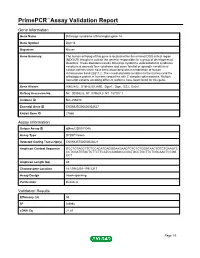
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DiGeorge syndrome critical region gene 14 Gene Symbol Dgcr14 Organism Mouse Gene Summary The human ortholog of this gene is located within the minimal DGS critical region (MDGCR) thought to contain the gene(s) responsible for a group of developmental disorders. These disorders include DiGeorge syndrome velocardiofacial syndrome conotruncal anomaly face syndrome and some familial or sporadic conotruncal cardiac defects which have been associated with microdeletion of human chromosome band 22q11.2. The encoded protein localizes to the nucleus and the orthologous protein in humans co-purifies with C complex spliceosomes. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases AI462402, D16H22S1269E, Dgcr1, Dgsi, ES2, Es2el RefSeq Accession No. NC_000082.6, NT_039624.8, NT_187007.1 UniGene ID Mm.256480 Ensembl Gene ID ENSMUSG00000003527 Entrez Gene ID 27886 Assay Information Unique Assay ID qMmuCID0011048 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000003621 Amplicon Context Sequence GCCTGTAGCTTCTCCACATCAGGGAAGAAGTCTCTCTGGATAACTGTCTGAAGTC CCTCGATGTACTCTTCTTCATCCAGGACCCGCTGCCTGCTTCTCGCAACTCCGG CCT Amplicon Length (bp) 82 Chromosome Location 16:17910201-17911217 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9994 cDNA Cq 21.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79.5 gDNA Cq 23.94 Specificity (%) 100 Information to assist with data interpretation is provided -

Parallel Next Generation Sequencing of DNA and RNA from a Single
www.nature.com/scientificreports OPEN Towards Improving Embryo Prioritization: Parallel Next Generation Sequencing of DNA and Received: 2 August 2018 Accepted: 14 January 2019 RNA from a Single Trophectoderm Published: xx xx xxxx Biopsy Noga Fuchs Weizman1, Brandon A. Wyse1, Ran Antes1, Zenon Ibarrientos1, Mugundhine Sangaralingam1, Gelareh Motamedi1, Valeriy Kuznyetsov1, Svetlana Madjunkova1 & Cliford L. Librach1,2,3,4 Improved embryo prioritization is crucial in optimizing the results in assisted reproduction, especially in light of increasing utilization of elective single embryo transfers. Embryo prioritization is currently based on morphological criteria and in some cases incorporates preimplantation genetic testing for aneuploidy (PGT-A). Recent technological advances have enabled parallel genomic and transcriptomic assessment of a single cell. Adding transcriptomic analysis to PGT-A holds promise for better understanding early embryonic development and implantation, and for enhancing available embryo prioritization tools. Our aim was to develop a platform for parallel genomic and transcriptomic sequencing of a single trophectoderm (TE) biopsy, that could later be correlated with clinical outcomes. Twenty-fve embryos donated for research were utilized; eight for initial development and optimization of our method, and seventeen to demonstrate clinical safety and reproducibility of this method. Our method achieved 100% concordance for ploidy status with that achieved by the classic PGT-A. All sequencing data exceeded quality control metrics. Transcriptomic sequencing data was sufcient for performing diferential expression (DE) analysis. All biopsies expressed specifc TE markers, further validating the accuracy of our method. Using PCA, samples clustered in euploid and aneuploid aggregates, highlighting the importance of controlling for ploidy in every transcriptomic assessment. -

CRNKL1 Is a Highly Selective Regulator of Intron-Retaining HIV-1 and Cellular Mrnas
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 CRNKL1 is a highly selective regulator of intron-retaining HIV-1 and cellular mRNAs 2 3 4 Han Xiao1, Emanuel Wyler2#, Miha Milek2#, Bastian Grewe3, Philipp Kirchner4, Arif Ekici4, Ana Beatriz 5 Oliveira Villela Silva1, Doris Jungnickl1, Markus Landthaler2,5, Armin Ensser1, and Klaus Überla1* 6 7 1 Institute of Clinical and Molecular Virology, University Hospital Erlangen, Friedrich-Alexander 8 Universität Erlangen-Nürnberg, Erlangen, Germany 9 2 Berlin Institute for Medical Systems Biology, Max-Delbrück-Center for Molecular Medicine in the 10 Helmholtz Association, Robert-Rössle-Strasse 10, 13125, Berlin, Germany 11 3 Department of Molecular and Medical Virology, Ruhr-University, Bochum, Germany 12 4 Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander Universität Erlangen- 13 Nürnberg, Erlangen, Germany 14 5 IRI Life Sciences, Institute für Biologie, Humboldt Universität zu Berlin, Philippstraße 13, 10115, Berlin, 15 Germany 16 # these two authors contributed equally 17 18 19 *Corresponding author: 20 Prof. Dr. Klaus Überla 21 Institute of Clinical and Molecular Virology, University Hospital Erlangen 22 Friedrich-Alexander Universität Erlangen-Nürnberg 23 Schlossgarten 4, 91054 Erlangen 24 Germany 25 Tel: (+49) 9131-8523563 26 e-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

C2orf3 (GCFC2) (NM 001201334) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG234563 C2orf3 (GCFC2) (NM_001201334) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: C2orf3 (GCFC2) (NM_001201334) Human Tagged ORF Clone Tag: TurboGFP Symbol: GCFC2 Synonyms: C2orf3; DNABF; GCF; TCF9 Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 C2orf3 (GCFC2) (NM_001201334) Human Tagged ORF Clone – RG234563 ORF Nucleotide >RG234563 representing NM_001201334 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGAAGAGAGAGAGCGAAGATGACCCTGAGAGTGAGCCTGATGACCATGAAAAGAGAATACCATTTACTC TAAGACCTCAAACACTTAGACAAAGGATGGCTGAGGAATCAATAAGCAGAAATGAAGAAACAAGTGAAGA AAGTCAGGAAGATGAAAAGCAAGATACTTGGGAACAACAGCAAATGAGGAAAGCAGTTAAAATCATAGAG GAAAGAGACATAGATCTTTCCTGTGGCAATGGATCTTCAAAAGTGAAGAAATTTGATACTTCCATTTCAT TTCCGCCAGTAAATTTAGAAATTATAAAGAAGCAATTAAATACTAGATTAACATTACTACAGGAAACTCA CCGCTCACACCTGAGGGAGTATGAAAAATACGTACAAGATGTCAAAAGCTCAAAGAGTACCATCCAGAAC CTAGAGAGTTCATCAAATCAAGCTCTAAATTGTAAATTCTATAAAAGCATGAAAATTTATGTGGAAAATT TAATTGACTGCCTTAATGAAAAGATTATCAACATCCAAGAAATAGAATCATCCATGCATGCACTCCTTTT -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Table SI. Genes Upregulated ≥ 2-Fold by MIH 2.4Bl Treatment Affymetrix ID
Table SI. Genes upregulated 2-fold by MIH 2.4Bl treatment Fold UniGene ID Description Affymetrix ID Entrez Gene Change 1558048_x_at 28.84 Hs.551290 231597_x_at 17.02 Hs.720692 238825_at 10.19 93953 Hs.135167 acidic repeat containing (ACRC) 203821_at 9.82 1839 Hs.799 heparin binding EGF like growth factor (HBEGF) 1559509_at 9.41 Hs.656636 202957_at 9.06 3059 Hs.14601 hematopoietic cell-specific Lyn substrate 1 (HCLS1) 202388_at 8.11 5997 Hs.78944 regulator of G-protein signaling 2 (RGS2) 213649_at 7.9 6432 Hs.309090 serine and arginine rich splicing factor 7 (SRSF7) 228262_at 7.83 256714 Hs.127951 MAP7 domain containing 2 (MAP7D2) 38037_at 7.75 1839 Hs.799 heparin binding EGF like growth factor (HBEGF) 224549_x_at 7.6 202672_s_at 7.53 467 Hs.460 activating transcription factor 3 (ATF3) 243581_at 6.94 Hs.659284 239203_at 6.9 286006 Hs.396189 leucine rich single-pass membrane protein 1 (LSMEM1) 210800_at 6.7 1678 translocase of inner mitochondrial membrane 8 homolog A (yeast) (TIMM8A) 238956_at 6.48 1943 Hs.741510 ephrin A2 (EFNA2) 242918_at 6.22 4678 Hs.319334 nuclear autoantigenic sperm protein (NASP) 224254_x_at 6.06 243509_at 6 236832_at 5.89 221442 Hs.374076 adenylate cyclase 10, soluble pseudogene 1 (ADCY10P1) 234562_x_at 5.89 Hs.675414 214093_s_at 5.88 8880 Hs.567380; far upstream element binding protein 1 (FUBP1) Hs.707742 223774_at 5.59 677825 Hs.632377 small nucleolar RNA, H/ACA box 44 (SNORA44) 234723_x_at 5.48 Hs.677287 226419_s_at 5.41 6426 Hs.710026; serine and arginine rich splicing factor 1 (SRSF1) Hs.744140 228967_at 5.37 -

Structural and Biochemical Studies of the Human DEAD-Box Helicase Dbp5 and Nucleoporin Nup214 Involved in Mrna Export‟ (Grade: 1.0) Since Sep
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Structural and Biochemical Studies of the Human DEAD-box Helicase Dbp5 and Nucleoporin Nup214 Involved in mRNA Export Holger von Moeller aus Leverkusen 2009 Erklärung Diese Dissertation wurde im Sinne von §13 Abs. 3 der Promotionsordnung vom 29. Januar 1998 von Frau Prof. Dr. Elena Conti betreut. Ehrenwörtliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, am 28.05.2009 Holger von Moeller Dissertation eingereicht am: 29.05.2009 1. Gutachter Prof. Dr. Elena Conti 2. Gutachter Prof. Dr. Patrick Cramer Mündliche Prüfung am: 30.09.2009 Meinen Eltern Abstract The hallmark of eukaryotic evolution was the development of the nucleus in cells. This compartmentalization requires the nucleocytoplasmic transport of thousands of molecules. The gate into and out of the nucleus is the nuclear pore complex (NPC). One of the molecules that needs to be exported from the nucleus is messenger RNA (mRNA). mRNA associates with proteins in the nucleus forming a messenger ribonucleoprotein particle (mRNP). mRNPs bind to dedicated transport factors that facilitate movement through the NPC. One protein that associates to mRNPs is the helicase Dbp5, which belongs to the DEAD-box family of RNA helicases. Dbp5 is essential for mRNA export in both yeast and humans. It binds RNA and is concentrated and locally activated at the cytoplasmic side of the nuclear pore complex, where it interacts with the cytoplasmic nucleoporin Nup214. In my PhD work, I have determined the crystal structures of human Dbp5 bound to RNA and AMPPNP, and bound to Nup214.