The Difficulty of Avoiding False Positives in Genome Scans for Natural Selection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

KRBA1 CRISPR/Cas9 KO Plasmid (M): Sc-429589
SANTA CRUZ BIOTECHNOLOGY, INC. KRBA1 CRISPR/Cas9 KO Plasmid (m): sc-429589 BACKGROUND APPLICATIONS The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and KRBA1 CRISPR/Cas9 KO Plasmid (m) is recommended for the disruption of CRISPR-associated protein (Cas9) system is an adaptive immune response gene expression in mouse cells. defense mechanism used by archea and bacteria for the degradation of for- eign genetic material (4,6). This mechanism can be repurposed for other 20 nt non-coding RNA sequence: guides Cas9 functions, including genomic engineering for mammalian systems, such as to a specific target location in the genomic DNA gene knockout (KO) (1,2,3,5). CRISPR/Cas9 KO Plasmid products enable the U6 promoter: drives gRNA scaffold: helps Cas9 identification and cleavage of specific genes by utilizing guide RNA (gRNA) expression of gRNA bind to target DNA sequences derived from the Genome-scale CRISPR Knock-Out (GeCKO) v2 library developed in the Zhang Laboratory at the Broad Institute (3,5). Termination signal Green Fluorescent Protein: to visually REFERENCES verify transfection CRISPR/Cas9 Knockout Plasmid CBh (chicken β-Actin 1. Cong, L., et al. 2013. Multiplex genome engineering using CRISPR/Cas hybrid) promoter: drives systems. Science 339: 819-823. 2A peptide: expression of Cas9 allows production of both Cas9 and GFP from the 2. Mali, P., et al. 2013. RNA-guided human genome engineering via Cas9. same CBh promoter Science 339: 823-826. Nuclear localization signal 3. Ran, F.A., et al. 2013. Genome engineering using the CRISPR-Cas9 system. Nuclear localization signal SpCas9 ribonuclease Nat. Protoc. 8: 2281-2308. -

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Topological Scoring of Protein Interaction Networks
bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Topological Scoring of Protein Interaction Networks Mihaela E. Sardiu1, Joshua M. Gilmore1,2, Brad D. Groppe1,3, Arnob Dutta1,4, Laurence Florens1, and Michael P. Washburn1,5‡ 1Stowers Institute for Medical Research, Kansas City, MO 64110 U.S.A. 2Current Address: Boehringer Ingelheim Vetmedica, St. Joseph, MO 64506 U.S.A. 3Current Address: Thermo Fisher Scientific, Waltham, MA 02451, U.S.A. 4Current Address: Department of Cell and Molecular Biology, University of Rhode Island, 287 CBLS, 120 Flagg Road, Kingston, RI 02881. 5 Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, Kansas 66160, USA ‡To whom correspondence should be addressed: Michael Washburn, Ph.D. Stowers Institute for Medical Research 1000 E. 50th St. Kansas City, MO 64110 Phone: 816-926-4457 E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract It remains a significant challenge to define individual protein associations within networks where an individual protein can directly interact with other proteins and/or be part of large complexes, which contain functional modules. Here we demonstrate the topological scoring (TopS) algorithm for the analysis of quantitative proteomic analyses of affinity purifications. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
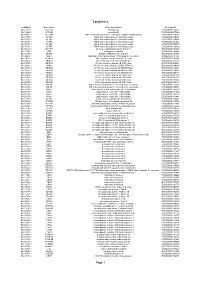
Epigenetics Page 1
Epigenetics esiRNA ID Gene Name Gene Description Ensembl ID HU-13237-1 ACTL6A actin-like 6A ENSG00000136518 HU-13925-1 ACTL6B actin-like 6B ENSG00000077080 HU-14457-1 ACTR1A ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) ENSG00000138107 HU-10579-1 ACTR2 ARP2 actin-related protein 2 homolog (yeast) ENSG00000138071 HU-10837-1 ACTR3 ARP3 actin-related protein 3 homolog (yeast) ENSG00000115091 HU-09776-1 ACTR5 ARP5 actin-related protein 5 homolog (yeast) ENSG00000101442 HU-00773-1 ACTR6 ARP6 actin-related protein 6 homolog (yeast) ENSG00000075089 HU-07176-1 ACTR8 ARP8 actin-related protein 8 homolog (yeast) ENSG00000113812 HU-09411-1 AHCTF1 AT hook containing transcription factor 1 ENSG00000153207 HU-15150-1 AIRE autoimmune regulator ENSG00000160224 HU-12332-1 AKAP1 A kinase (PRKA) anchor protein 1 ENSG00000121057 HU-04065-1 ALG13 asparagine-linked glycosylation 13 homolog (S. cerevisiae) ENSG00000101901 HU-13552-1 ALKBH1 alkB, alkylation repair homolog 1 (E. coli) ENSG00000100601 HU-06662-1 ARID1A AT rich interactive domain 1A (SWI-like) ENSG00000117713 HU-12790-1 ARID1B AT rich interactive domain 1B (SWI1-like) ENSG00000049618 HU-09415-1 ARID2 AT rich interactive domain 2 (ARID, RFX-like) ENSG00000189079 HU-03890-1 ARID3A AT rich interactive domain 3A (BRIGHT-like) ENSG00000116017 HU-14677-1 ARID3B AT rich interactive domain 3B (BRIGHT-like) ENSG00000179361 HU-14203-1 ARID3C AT rich interactive domain 3C (BRIGHT-like) ENSG00000205143 HU-09104-1 ARID4A AT rich interactive domain 4A (RBP1-like) ENSG00000032219 HU-12512-1 ARID4B AT rich interactive domain 4B (RBP1-like) ENSG00000054267 HU-12520-1 ARID5A AT rich interactive domain 5A (MRF1-like) ENSG00000196843 HU-06595-1 ARID5B AT rich interactive domain 5B (MRF1-like) ENSG00000150347 HU-00556-1 ASF1A ASF1 anti-silencing function 1 homolog A (S. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Supp Material.Pdf
Supplementary Information Estrogen-mediated Epigenetic Repression of Large Chromosomal Regions through DNA Looping Pei-Yin Hsu, Hang-Kai Hsu, Gregory A. C. Singer, Pearlly S. Yan, Benjamin A. T. Rodriguez, Joseph C. Liu, Yu-I Weng, Daniel E. Deatherage, Zhong Chen, Julia S. Pereira, Ricardo Lopez, Jose Russo, Qianben Wang, Coral A. Lamartiniere, Kenneth P. Nephew, and Tim H.-M. Huang S1 Method Immunofluorescence staining Approximately 2,000 mammosphere-derived epithelial cells (MDECs) cells seeded collagen I-coated coverslips were fixed with methanol/acetone for 10 min. After blocking with 2.5% bovine serum albumin (Sigma) for 1 hr, these cells were incubated with anti-ESR1 antibody (Santa Cruz) overnight at 4˚C. The corresponding secondary FITC-conjugated antibody was applied followed by DAPI staining (Molecular Probes) for the nuclei. Photographs were captured by Zeiss fluorescence microscopy (Zeiss). The percentages of ESR1 subcellular localization were calculated in ten different optical fields (~10 cells per field) by two independent researchers. References Carroll, J.S., Meyer, C.A., Song, J., Li, W., Geistlinger, T.R., Eeckhoute, J., Brodsky, A.S., Keeton, E.K., Fertuck, K.C., Hall, G.F., et al. 2006. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38: 1289-1297. Neve, R.M., Chin, K., Fridlyand, J., Yeh, J., Baehner, F.L., Fevr, T., Clark, L., Bayani, N., Coppe, J.P., Tong, F., et al. 2006. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10: 515-527. S2 Hsu et al. Supplementary Information A Figure S1. Integrative mapping of large genomic regions subjected to ERα-mediated epigenetic repression. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Supp Material.Pdf
Simon et al. Supplementary information: Table of contents p.1 Supplementary material and methods p.2-4 • PoIy(I)-poly(C) Treatment • Flow Cytometry and Immunohistochemistry • Western Blotting • Quantitative RT-PCR • Fluorescence In Situ Hybridization • RNA-Seq • Exome capture • Sequencing Supplementary Figures and Tables Suppl. items Description pages Figure 1 Inactivation of Ezh2 affects normal thymocyte development 5 Figure 2 Ezh2 mouse leukemias express cell surface T cell receptor 6 Figure 3 Expression of EZH2 and Hox genes in T-ALL 7 Figure 4 Additional mutation et deletion of chromatin modifiers in T-ALL 8 Figure 5 PRC2 expression and activity in human lymphoproliferative disease 9 Figure 6 PRC2 regulatory network (String analysis) 10 Table 1 Primers and probes for detection of PRC2 genes 11 Table 2 Patient and T-ALL characteristics 12 Table 3 Statistics of RNA and DNA sequencing 13 Table 4 Mutations found in human T-ALLs (see Fig. 3D and Suppl. Fig. 4) 14 Table 5 SNP populations in analyzed human T-ALL samples 15 Table 6 List of altered genes in T-ALL for DAVID analysis 20 Table 7 List of David functional clusters 31 Table 8 List of acquired SNP tested in normal non leukemic DNA 32 1 Simon et al. Supplementary Material and Methods PoIy(I)-poly(C) Treatment. pIpC (GE Healthcare Lifesciences) was dissolved in endotoxin-free D-PBS (Gibco) at a concentration of 2 mg/ml. Mice received four consecutive injections of 150 μg pIpC every other day. The day of the last pIpC injection was designated as day 0 of experiment. -

A Dissertation Entitled the Androgen Receptor
A Dissertation entitled The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit Submitted to the Graduate Faculty as partial fulfillment of the requirements for the PhD Degree in Biomedical science Dr. Manohar Ratnam, Committee Chair Dr. Lirim Shemshedini, Committee Member Dr. Robert Trumbly, Committee Member Dr. Edwin Sanchez, Committee Member Dr. Beata Lecka -Czernik, Committee Member Dr. Patricia R. Komuniecki, Dean College of Graduate Studies The University of Toledo August 2011 Copyright 2011, Mesfin Gonit This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of The Androgen Receptor as a Transcriptional Co-activator: Implications in the Growth and Progression of Prostate Cancer By Mesfin Gonit As partial fulfillment of the requirements for the PhD Degree in Biomedical science The University of Toledo August 2011 Prostate cancer depends on the androgen receptor (AR) for growth and survival even in the absence of androgen. In the classical models of gene activation by AR, ligand activated AR signals through binding to the androgen response elements (AREs) in the target gene promoter/enhancer. In the present study the role of AREs in the androgen- independent transcriptional signaling was investigated using LP50 cells, derived from parental LNCaP cells through extended passage in vitro. LP50 cells reflected the signature gene overexpression profile of advanced clinical prostate tumors. The growth of LP50 cells was profoundly dependent on nuclear localized AR but was independent of androgen. Nevertheless, in these cells AR was unable to bind to AREs in the absence of androgen. -

Accumulated Degeneration of Transcriptional Regulation Contributes to Disease Development and Detrimental Clinical Outcomes of Alzheimer’S Disease
bioRxiv preprint doi: https://doi.org/10.1101/779249; this version posted September 28, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Accumulated degeneration of transcriptional regulation contributes to disease development and detrimental clinical outcomes of Alzheimer's disease Guofeng Meng1,*, Dong Lu1, Feng Yu1, Jijia Sun1, Chong Ding1, Yan Sun2, Xuan Liu1, Jiapei Dai2, Wenfei jin3, and Weidong Zhang1,* 1Institute of interdisciplinary integrative Medicine Research, shanghai University of Traditional Chinese Medicine, shanghai, China 2Wuhan Institute for Neuroscience and Neuroengineering (WINN), South-Central University for Nationalities, Wuhan 430074, China 3Department of Biology, Southwest University of Science and Technology, Shenzhen, China *Correspondence: [email protected] and [email protected] Abstract Alzheimer's disease (AD) is extremely complex for both causal mechanism and clinical manifestation, requiring efforts to uncover its diversity and the corresponding mechanisms. Here, we applied a modelling analysis to investigate the regulation divergence among a large-scale cohort of AD patients. We found that transcription regulation tended to get degenerated in AD patients, which contributed to disease development and the detrimental clinical outcomes, mainly by disrupting protein degradation, neuroinflammation, mitochondrial and synaptic functions. To measure the accumulated effects,we came up with a new concept, regulation loss burden, which better correlated with AD related clinical manifestations and the ageing process. The epigenetic studies to multiple active regulation marks also supported a tendency of regulation loss in AD patients.