1.01 Aziridines and Azirines: Monocyclic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

New Methods for the Synthesis of Heterocyclic Compounds*
Pure Appl. Chem., Vol. 76, No. 3, pp. 603–613, 2004. © 2004 IUPAC New methods for the synthesis of heterocyclic compounds* Aldo Caiazzo1, Shadi Dalili1, Christine Picard2, Mikio Sasaki1, Tung Siu2, and Andrei K. Yudin1,‡ 1Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada; 2Ylektra, Inc., 100 University Ave., 10th Floor, South Tower, Toronto, Ontario M5J 1V6, Canada Abstract: Due to frequent occurrence of nitrogen-containing groups among the biologically active compounds, chemoselective functionalization of organic molecules with nitrogen-con- taining functional groups is an important area of organic synthesis. We have proposed and implemented a new strategy toward design of nitrogen-transfer reactions on inert electrode surfaces with a particular focus on the generation and trapping of highly reactive nitrogen- transfer agents. A wide range of structurally dissimilar olefins can be readily transformed into the corresponding aziridines. The resulting aziridines are precursors to a range of cata- lysts via nucleophilic ring-opening with diaryl- and dialkyl phosphines. Another strategy ex- plored in the context of oxidative nitrogen transfer is cycloamination of olefins using NH aziridines. INTRODUCTION The transformations of organic compounds belong to one of the following two broad categories: car- bon–carbon bond-forming reactions and redox processes. Over the years, remarkable progress has been achieved in design and applications of novel metal-based complexes in oxidation chemistry. The metal center of such a catalyst is surrounded by a ligand, which resembles and emulates the function of the enzyme active site. This strategy is known to adequately address the issues of regio-, chemo-, and stereoselectivity in a number of synthetic transformations. -

Recent Advances in Titanium Radical Redox Catalysis
JOCSynopsis Cite This: J. Org. Chem. 2019, 84, 14369−14380 pubs.acs.org/joc Recent Advances in Titanium Radical Redox Catalysis Terry McCallum, Xiangyu Wu, and Song Lin* Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States ABSTRACT: New catalytic strategies that leverage single-electron redox events have provided chemists with useful tools for solving synthetic problems. In this context, Ti offers opportunities that are complementary to late transition metals for reaction discovery. Following foundational work on epoxide reductive functionalization, recent methodological advances have significantly expanded the repertoire of Ti radical chemistry. This Synopsis summarizes recent developments in the burgeoning area of Ti radical catalysis with a focus on innovative catalytic strategies such as radical redox-relay and dual catalysis. 1. INTRODUCTION a green chemistry perspective, the abundance and low toxicity of Ti make its complexes highly attractive as reagents and Radical-based chemistry has long been a cornerstone of 5 1 catalysts in organic synthesis. synthetic organic chemistry. The high reactivity of organic IV/III radicals has made possible myriad new reactions that cannot be A classic example of Ti -mediated reactivity is the reductive ring opening of epoxides. This process preferentially readily achieved using two-electron chemistry. However, the − high reactivity of organic radicals is a double-edged sword, as cleaves and functionalizes the more substituted C O bond, the selectivity of these fleeting intermediates can be difficult to providing complementary regioselectivity to Lewis acid control in the presence of multiple chemotypes. In addition, promoted epoxide reactions. The synthetic value of Ti redox catalysis has been highlighted by their many uses in total catalyst-controlled regio- and stereoselective reactions involv- 6−10 ing free-radical intermediates remain limited,2 and the synthesis (Scheme 1). -

The Synthesis and Applications of N-Alkenyl Aziridines
The Synthesis and Applications of N-Alkenyl Aziridines by Nicholas A. Afagh A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Nicholas A. Afagh 2010 The Synthesis and Applications of N-Alkenyl Aziridines Nicholas A. Afagh Master of Science Department of Chemistry University of Toronto 2010 Abstract N-alkenyl aziridines are a unique class of molecules that do not behave as typical enamines as a result of the inability of the nitrogen atom lone-pair of electrons to delocalize. The attenuated nucleophilicity of these enamines presents opportunities for the selective functionalization and reactivity not available to classical enamines. An operationally simple and mild copper-mediated coupling has been developed that facilitates the preparation of a broad range of N-alkenyl aziridines not available through existing methods. The preparation and reactivity of highly- functionalized N-alkenyl aziridines are reported. Also reported is the application of the chemoselective amine/aldehyde/alkyne (A 3) multicomponent coupling involving amphoteric aziridine aldehydes as the aldehyde component. This coupling allows access to propargyl amines with pendent aziridine functionality. ii Acknowledgments First and foremost, I would like to thank my supervisor, Professor Andrei K. Yudin for his continuous support and encouragement over the past two years. His wealth of knowledge and profound insight into all matters chemistry made for many interesting discussions. In addition, I would like to thank all the members of the Yudin group past and present with whom I have had the distinct pleasure of working alongside and shared many late evenings. -

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

Aziridination of Alkenes Promoted by Iron Or Ruthenium Complexes
Aziridination of Alkenes Promoted by Iron or Ruthenium Complexes Caterina Damiano, Daniela Intrieri and Emma Gallo* Department of Chemistry, University of Milan, Via C. Golgi 19, 20133 Milan (Italy). E-mail address: [email protected]. Keywords: Aziridines, Nitrene reagents, Alkenes, Homogenous catalysis, Iron, Ruthenium. Abstract Molecules containing an aziridine functional group are a versatile class of organic synthons due to the presence of a strained three member, which can be easily involved in ring-opening reactions and the aziridine functionality often show interesting pharmaceutical and/or biological behaviours. For these reasons, the scientific community is constantly interested in developing efficient procedures to introduce an aziridine moiety into organic skeletons and the one-pot reaction of an alkene double bond with a nitrene [NR] source is a powerful synthetic strategy. Herein we describe the catalytic activity of iron or ruthenium complexes in promoting the reaction stated above by stressing the potential and limits of each synthetic protocol. 1. Introduction Aziridines, the smallest N-heterocycle compounds, have attracted considerable attention in the last few decades due to their many applications in biological and synthetic chemistry [1]. The aziridine functionality is often responsible for the activity of biologically active species (such as antitumor compounds, antibiotics and enzyme inhibitors) and aziridine containing molecules [2] are also useful building blocks in the synthesis of fine chemicals and pharmaceuticals [3-6]. The striking chemical properties of aziridines are due to the energy associated to the strained three- membered ring [7], which renders them very active and versatile starting materials for the synthesis of several useful molecules such as amines, amino acids, β-lactams, polymers and α-amido ketones [8, 9]. -

The Effect of High-Dose Thiotepa, Alone Or in Combination with Other
Bone Marrow Transplantation (2007) 40, 891–896 & 2007 Nature Publishing Group All rights reserved 0268-3369/07 $30.00 www.nature.com/bmt ORIGINAL ARTICLE The effect of high-dose thiotepa, alone or in combination with other chemotherapeutic agents, on a murine B-cell leukemia model simulating autologous stem cell transplantation A Abdul-Hai, L Weiss, D Ergas, IB Resnick, S Slavin and MY Shapira Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah–Hebrew University Medical Center, Jerusalem, Israel The use of thiotepa (TH) is increasing, especially in stem Introduction cell transplantation, mainly due to its safety and blood– brain barrier penetration. We evaluated the use of TH in a Thiotepa (TH, triethylene thiophosphoramide), an ethylene murine model simulating autologous stem cell transplan- amide, developed by Lederle Laboratories in 1952, tation, with or without additional agents. Between 1 and possesses mechlorethamine-like alkylating activity and 11 days following inoculation of BALB/c mice with 105– has been used clinically for over 35 years.1 It is of particular 108 B-cell leukemia (BCL1) cells (simulating pre-trans- use in breast cancer, mainly as second-line treatment.2 plant leukemia loads), each group received an ‘induction- TH has been given intrathecally for carcinomatous like’ irradiation and/or cytotoxic regimen. Animals were meningitis, and intravesically for bladder carcinoma.3,4 either followed without treatment, or an adoptive transfer Mild-to-moderate activity was observed in several solid (AT) was performed to untreated BALB/c mice. Admi- tumors and hematological malignancies.1,5 There has nistered alone without AT, high-dose TH did not change been, however, a sustained interest in defining clinical roles the time to appearance of leukemia. -

Sigmatropic Rearrangement of Vinyl Aziridines: Expedient Synthesis of Cyclic Sulfoximines from Chiral Sulfinimines Toni Moragas,A Ryan M
Sigmatropic Rearrangement of Vinyl Aziridines: Expedient Synthesis of Cyclic Sulfoximines from Chiral Sulfinimines Toni Moragas,a Ryan M. Liffey,a Dominika Regentová,a Jon-Paul S. Ward,a Justine Dutton,a William Lewis,a Ian Churcher,b Lesley Walton,c José A. Souto,a,d Robert A. Stockmana* Abstract: A novel rearrangement of 2-vinyl aziridine 2-carboxylates to unusual chiral cyclic sulfoximines is described herein. The method allows the synthesis of substituted cyclic sulfoximines in high yields with complete stereocontrol, displaying a wide substrate scope under mild conditions. Further development of a one-pot process directly from sulfinimines shows the synthetic applicability of this protocol, providing access to complex chiral sulfoximines in only two steps from commercially available aldehydes. A mechanistic hypothesis and synthetic application in the formal synthesis of trachelanthamidine by transformation of a cyclic sulfoximine into a pyrroline is also disclosed. Since the first isolation of sulfoximines by Bentley and coworkers in 19501 these sulfur containing compounds have found applications in functional group transformations and asymmetric synthesis,2 drug development,3 crop treatment4 and insect control.5 Largely ignored in medicinal chemistry for around 50 years, the sulfoximine group has recently been the object of significant new interest in this area.3 Sulfoximines are three-dimensional motifs with three points of attachment in orthogonal vectors,6 with functionalisation at nitrogen and carbon alpha to the sulfur both versatile and facile. 7-9 Despite the promising biological activity showed by the few previously synthesised cyclic sulfoximines,3 methods describing the synthesis of these compounds are scarce and mainly involve the multistep synthesis of linear sulfoximines10,11 and subsequent cyclisations.12 Furthermore, they generally describe benzo-fused cyclic sulfoximines. -

SUMMARY of PARTICULARLY HAZARDOUS SUBSTANCES (By
SUMMARY OF PARTICULARLY HAZARDOUS SUBSTANCES (by alpha) Key: SC -- Select Carcinogens RT -- Reproductive Toxins AT -- Acute Toxins SA -- Readily Absorbed Through the Skin DHS -- Chemicals of Interest Revised: 11/2012 ________________________________________________________ ___________ _ _ _ _ _ _ _ _ _ _ _ ||| | | | CHEMICAL NAME CAS # |SC|RT| AT | SA |DHS| ________________________________________________________ ___________ | _ | _ | _ | _ | __ | | | | | | | 2,4,5-T 000093-76-5 | | x | | x | | ABRIN 001393-62-0 | | | x | | | ACETALDEHYDE 000075-07-0 | x | | | | | ACETAMIDE 000060-35-5 | x | | | | | ACETOHYDROXAMIC ACID 000546-88-3 ||x| | x | | ACETONE CYANOHYDRIN, STABILIZED 000075-86-5 | | | x | | x | ACETYLAMINOFLUORENE,2- 000053-96-3 | x | | | | | ACID MIST, STRONG INORGANIC 000000-00-0 | x | | | | | ACROLEIN 000107-02-8 | | x | x | x | | ACRYLAMIDE 000079-06-1 | x | x | | x | | ACRYLONITRILE 000107-13-1 | x | x | x | x | | ACTINOMYCIN D 000050-76-0 ||x| | x | | ADIPONITRILE 000111-69-3 | | | x | | | ADRIAMYCIN 023214-92-8 | x | | | | | AFLATOXIN B1 001162-65-8 | x | | | | | AFLATOXIN M1 006795-23-9 | x | | | | | AFLATOXINS 001402-68-2 | x | | x | | | ALL-TRANS RETINOIC ACID 000302-79-4 | | x | | x | | ALPRAZOMAN 028981-97-7 | | x | | x | | ALUMINUM PHOSPHIDE 020859-73-8 | | | x | | x | AMANTADINE HYDROCHLORIDE 000665-66-7 | | x | | x | | AMINO-2,4-DIBROMOANTHRAQUINONE 000081-49-2 | x | | | | | AMINO-2-METHYLANTHRAQUINONE, 1- 000082-28-0 | x | | | | | AMINO-3,4-DIMETHYL-3h-IMIDAZO(4,5f)QUINOLINE,2- 077094-11-2 | x | | | | | AMINO-3,8-DIMETHYL-3H-IMIDAZO(4,5-f)QUINOXALINE, -

Electrocyclic Reactions of Aziridines
ELECTROCYCLIC RUCTIONS OF AZIRIDINES by MUHAMMAD HUMAYOUN AKHTAR M. Sc., University of Moncton, 1966. A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in the Department of Chemistry @ WHAMMAD HUMAYOUN AKHTAR , 1970 SIMON FRASER UNIVERSITY August, 1970 A PPR OVA L Name : Muhammad Humayoun Akhtar Degree : Doc tor of Philosophy Title of' Thesis: Electrocyclic Reactions of Aziridines Examining Committee: ,, A. C . Oehlschlaire'u' Senior supervisa/ . .- ' A. G. SherviooP Examining Committee - - K. ?I.Slessor ~xami~kn~Committee A. Fischer External Examiner Professor Department of Chemistry The University of Victoria Victoria, British Columbia Abstract Temperature dependent nmr spectra have been observed for a series of para-substi tuted 1-aryl-3,3- dimethyltriazenes . The temperature dependence has been interpreted in terms of restricted internal rotation about the N2,Fj bond of these triazenes and activation para- 5 meters AF', AH', and AS have been calculated from the spectral data. The origin of the rotational barrier is considered to lie in partial double bond formation between hT2 and T3 due to the contribution of 1,3-dipolar resonance hybrids to the qround states of these triazenes. This interpretation is supported by a sizable substituent effect o= -2'1 for the rotational process in the series studied. Chemical shift data at low temperatures indicates a stereo- specific association of benzene with the triazenes studied which places the benzene ring closer to the trans N-Me group in the 1,3-dipolar resonance hybrid. The thermal decomposition of ~T-arylazoaziridines follows two routes; one giving arylazide and alkene (stereospecif ically) and the other giving products typical of homolysis of the azo-linkage. -

Asymmetric Nitrene Transfer Reactions with Azides Via Co(II)-Based Metalloradical Catalysis (MRC) Jingran Tao University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2013 Asymmetric Nitrene Transfer Reactions with Azides via Co(II)-Based Metalloradical Catalysis (MRC) Jingran Tao University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Chemistry Commons Scholar Commons Citation Tao, Jingran, "Asymmetric Nitrene Transfer Reactions with Azides via Co(II)-Based Metalloradical Catalysis (MRC)" (2013). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4590 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Asymmetric Nitrene Transfer Reactions with Azides via Co(II)-Based Metalloradical Catalysis (MRC) by Jingran Tao A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Chemistry College of Arts and Sciences University of South Florida Major Professor: X. Peter Zhang, Ph.D. Jon Antilla, Ph.D. Wayne Guida, Ph.D. Xiao Li, Ph.D. Date of Approval: April 3rd , 2013 Keywords: cobalt, porphyrin, catalysis, aziridination, C–H amination, azide, asymmetric Copyright © 2013, Jingran Tao Dedication I dedicate this dissertation to my beloved parents. Acknowledgments I need to begin with thanking Dr. Peter Zhang for his continuous guidance and support. I learned the words “determination” and “believe” from him. I also need to thank my committee members: Dr. Jon Antilla, Dr. Wayne Guida Dr. Xiao Li and Chair Dr. -
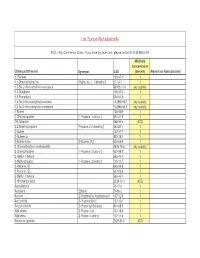
Homeland Security List
List Sorted Alphabetically ACG = Any Commercial Grade. If you have any questions, please contact EHS @ 898-5126. Minimum Concentration Chemical Of Interest Synonym CAS (percent) Amount on Hand (pounds) 1- Pentene 109-67-1 1 1,1-Dimethylhydrazine [Hydrazine, 1, 1-dimethyl-] 57-14-7 1 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 any quantity 1,3-Butadiene 106-99-0 1 1,3-Pentadiene 504-60-9 1 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 any quantity 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 any quantity 1-Butene 106-98-9 1 1-Chloropropylene [1-Propene, 1-chloro-] 590-21-6 1 1H-Tetrazole 288-94-8 ACG 2,2-Dimethylpropane [Propane, 2,2-dimethyl-] 463-82-1 1 2-Butene 107-01-7 1 2-Butene-cis 590-18-1 1 2-Butene-trans [2-Butene, (E)] 624-64-6 1 2-Chloroethylchloro-methylsulfide 2625-76-5 any quantity 2-Chloropropylene [1-Propene, 2-chloro-] 557-98-2 1 2-Methyl-1-butene 563-46-2 1 2-Methylpropene [1-Propene, 2-methyl-] 115-11-7 1 2-Pentene, (E)- 646-04-8 1 2-Pentene, (Z)- 627-20-3 1 3-Methyl-1-butene 563-45-1 1 5-Nitrobenzotriazol 2338-12-7 ACG Acetaldehyde 75-07-0 1 Acetylene [Ethyne] 74-86-2 1 Acrolein [2-Propenal] or Acrylaldehyde 107-02-8 1 Acrylonitrile [2-Propenenitrile] 107-13-1 1 Acrylyl chloride [2-Propenoyl chloride] 814-68-6 1 Allyl alcohol [2-Propen-1-ol] 107-18-6 1 Allylamine [2-Propen-1-amine] 107-11-9 1 Aluminum (powder) 7429-90-5 ACG Ammonia (anhydrous) 7664-41-7 1 Ammonia (conc. -

1,3-Dipolar Cycloaddition of Azomethine Ylides Generated from Aziridines in Supercritical Carbon Dioxide Paulo J
Tetrahedron Letters 47 (2006) 5475–5479 1,3-Dipolar cycloaddition of azomethine ylides generated from aziridines in supercritical carbon dioxide Paulo J. S. Gomes, Cla´udio M. Nunes, Alberto A. C. C. Pais, Teresa M. V. D. Pinho e Melo* and Luis G. Arnaut Department of Chemistry, Coimbra University, 3004-535 Coimbra, Portugal Received 8 March 2006; revised 26 May 2006; accepted 30 May 2006 Abstract—The 1,3-dipolar cycloaddition of azomethine ylides with DMAD in supercritical carbon dioxide is reported. The photo- lysis reaction conditions were optimized with a suitable adjustment of pressure, temperature, irradiation time and co-solvent concen- tration leading to a more efficient reaction than in neat acetonitrile. Similar results were observed using thermal reaction conditions. Supercritical carbon dioxide with a minute co-solvent addition is shown to be a very efficient medium for promoting this type of cycloadditions. Ó 2006 Elsevier Ltd. All rights reserved. Over the last decade, supercritical fluids (SCF) received ology for the synthesis of nitrogen-heterocyclic significant attention as a new class of environmentally compounds. We chose the 1,3-dipolar cycloaddition friendly solvents for organic synthesis. Supercritical flu- of azomethine ylides generated from aziridines as the ids have properties intermediate between the gas and the model reaction. Aziridines (1) undergo electrocyclic ring liquid phases that can be tuned simply by changing the opening in a disrotatory manner upon irradiation and pressure and temperature. In particular, changes of pres- conrotatory ring opening upon thermolysis, giving sure close to the critical point enable drastic changes in azomethine ylides (2), which participate in 1,3-dipolar density and viscosity.