Taxize: Taxonomic Search and Retrieval in R[V2; Ref Status
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bulletin / New York State Museum
Juncaceae (Rush Family) of New York State Steven E. Clemants New York Natural Heritage Program LIBRARY JUL 2 3 1990 NEW YORK BOTANICAL GARDEN Contributions to a Flora of New York State VII Richard S. Mitchell, Editor Bulletin No. 475 New York State Museum The University of the State of New York THE STATE EDUCATION DEPARTMENT Albany, New York 12230 NEW YORK THE STATE OF LEARNING Digitized by the Internet Archive in 2017 with funding from IMLS LG-70-15-0138-15 https://archive.org/details/bulletinnewyorks4751 newy Juncaceae (Rush Family) of New York State Steven E. Clemants New York Natural Heritage Program Contributions to a Flora of New York State VII Richard S. Mitchell, Editor 1990 Bulletin No. 475 New York State Museum The University of the State of New York THE STATE EDUCATION DEPARTMENT Albany, New York 12230 THE UNIVERSITY OF THE STATE OF NEW YORK Regents of The University Martin C. Barell, Chancellor, B.A., I. A., LL.B Muttontown R. Carlos Carballada, Vice Chancellor , B.S Rochester Willard A. Genrich, LL.B Buffalo Emlyn 1. Griffith, A. B., J.D Rome Jorge L. Batista, B. A., J.D Bronx Laura Bradley Chodos, B.A., M.A Vischer Ferry Louise P. Matteoni, B.A., M.A., Ph.D Bayside J. Edward Meyer, B.A., LL.B Chappaqua Floyd S. Linton, A.B., M.A., M.P.A Miller Place Mimi Levin Lieber, B.A., M.A Manhattan Shirley C. Brown, B.A., M.A., Ph.D Albany Norma Gluck, B.A., M.S.W Manhattan James W. -

State of New York City's Plants 2018
STATE OF NEW YORK CITY’S PLANTS 2018 Daniel Atha & Brian Boom © 2018 The New York Botanical Garden All rights reserved ISBN 978-0-89327-955-4 Center for Conservation Strategy The New York Botanical Garden 2900 Southern Boulevard Bronx, NY 10458 All photos NYBG staff Citation: Atha, D. and B. Boom. 2018. State of New York City’s Plants 2018. Center for Conservation Strategy. The New York Botanical Garden, Bronx, NY. 132 pp. STATE OF NEW YORK CITY’S PLANTS 2018 4 EXECUTIVE SUMMARY 6 INTRODUCTION 10 DOCUMENTING THE CITY’S PLANTS 10 The Flora of New York City 11 Rare Species 14 Focus on Specific Area 16 Botanical Spectacle: Summer Snow 18 CITIZEN SCIENCE 20 THREATS TO THE CITY’S PLANTS 24 NEW YORK STATE PROHIBITED AND REGULATED INVASIVE SPECIES FOUND IN NEW YORK CITY 26 LOOKING AHEAD 27 CONTRIBUTORS AND ACKNOWLEGMENTS 30 LITERATURE CITED 31 APPENDIX Checklist of the Spontaneous Vascular Plants of New York City 32 Ferns and Fern Allies 35 Gymnosperms 36 Nymphaeales and Magnoliids 37 Monocots 67 Dicots 3 EXECUTIVE SUMMARY This report, State of New York City’s Plants 2018, is the first rankings of rare, threatened, endangered, and extinct species of what is envisioned by the Center for Conservation Strategy known from New York City, and based on this compilation of The New York Botanical Garden as annual updates thirteen percent of the City’s flora is imperiled or extinct in New summarizing the status of the spontaneous plant species of the York City. five boroughs of New York City. This year’s report deals with the City’s vascular plants (ferns and fern allies, gymnosperms, We have begun the process of assessing conservation status and flowering plants), but in the future it is planned to phase in at the local level for all species. -

Literature Cited
Literature Cited Robert W. Kiger, Editor This is a consolidated list of all works cited in volumes 19, 20, and 21, whether as selected references, in text, or in nomenclatural contexts. In citations of articles, both here and in the taxonomic treatments, and also in nomenclatural citations, the titles of serials are rendered in the forms recommended in G. D. R. Bridson and E. R. Smith (1991). When those forms are abbre- viated, as most are, cross references to the corresponding full serial titles are interpolated here alphabetically by abbreviated form. In nomenclatural citations (only), book titles are rendered in the abbreviated forms recommended in F. A. Stafleu and R. S. Cowan (1976–1988) and F. A. Stafleu and E. A. Mennega (1992+). Here, those abbreviated forms are indicated parenthetically following the full citations of the corresponding works, and cross references to the full citations are interpolated in the list alphabetically by abbreviated form. Two or more works published in the same year by the same author or group of coauthors will be distinguished uniquely and consistently throughout all volumes of Flora of North America by lower-case letters (b, c, d, ...) suffixed to the date for the second and subsequent works in the set. The suffixes are assigned in order of editorial encounter and do not reflect chronological sequence of publication. The first work by any particular author or group from any given year carries the implicit date suffix “a”; thus, the sequence of explicit suffixes begins with “b”. Works missing from any suffixed sequence here are ones cited elsewhere in the Flora that are not pertinent in these volumes. -

Nature Conservation
J. Nat. Conserv. 11, – (2003) Journal for © Urban & Fischer Verlag http://www.urbanfischer.de/journals/jnc Nature Conservation Constructing Red Numbers for setting conservation priorities of endangered plant species: Israeli flora as a test case Yuval Sapir1*, Avi Shmida1 & Ori Fragman1,2 1 Rotem – Israel Plant Information Center, Dept. of Evolution, Systematics and Ecology,The Hebrew University, Jerusalem, 91904, Israel; e-mail: [email protected] 2 Present address: Botanical Garden,The Hebrew University, Givat Ram, Jerusalem 91904, Israel Abstract A common problem in conservation policy is to define the priority of a certain species to invest conservation efforts when resources are limited. We suggest a method of constructing red numbers for plant species, in order to set priorities in con- servation policy. The red number is an additive index, summarising values of four parameters: 1. Rarity – The number of sites (1 km2) where the species is present. A rare species is defined when present in 0.5% of the area or less. 2. Declining rate and habitat vulnerability – Evaluate the decreasing rate in the number of sites and/or the destruction probability of the habitat. 3. Attractivity – the flower size and the probability of cutting or exploitation of the plant. 4. Distribution type – scoring endemic species and peripheral populations. The plant species of Israel were scored for the parameters of the red number. Three hundred and seventy (370) species, 16.15% of the Israeli flora entered into the “Red List” received red numbers above 6. “Post Mortem” analysis for the 34 extinct species of Israel revealed an average red number of 8.7, significantly higher than the average of the current red list. -
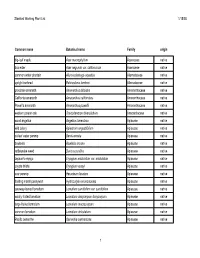
Plant List for Web Page
Stanford Working Plant List 1/15/08 Common name Botanical name Family origin big-leaf maple Acer macrophyllum Aceraceae native box elder Acer negundo var. californicum Aceraceae native common water plantain Alisma plantago-aquatica Alismataceae native upright burhead Echinodorus berteroi Alismataceae native prostrate amaranth Amaranthus blitoides Amaranthaceae native California amaranth Amaranthus californicus Amaranthaceae native Powell's amaranth Amaranthus powellii Amaranthaceae native western poison oak Toxicodendron diversilobum Anacardiaceae native wood angelica Angelica tomentosa Apiaceae native wild celery Apiastrum angustifolium Apiaceae native cutleaf water parsnip Berula erecta Apiaceae native bowlesia Bowlesia incana Apiaceae native rattlesnake weed Daucus pusillus Apiaceae native Jepson's eryngo Eryngium aristulatum var. aristulatum Apiaceae native coyote thistle Eryngium vaseyi Apiaceae native cow parsnip Heracleum lanatum Apiaceae native floating marsh pennywort Hydrocotyle ranunculoides Apiaceae native caraway-leaved lomatium Lomatium caruifolium var. caruifolium Apiaceae native woolly-fruited lomatium Lomatium dasycarpum dasycarpum Apiaceae native large-fruited lomatium Lomatium macrocarpum Apiaceae native common lomatium Lomatium utriculatum Apiaceae native Pacific oenanthe Oenanthe sarmentosa Apiaceae native 1 Stanford Working Plant List 1/15/08 wood sweet cicely Osmorhiza berteroi Apiaceae native mountain sweet cicely Osmorhiza chilensis Apiaceae native Gairdner's yampah (List 4) Perideridia gairdneri gairdneri Apiaceae -

New Zealand Rushes: Juncus Factsheets
New Zealand Rushes: Juncus factsheets K. Bodmin, P. Champion, T. James and T. Burton www.niwa.co.nz Acknowledgements: Our thanks to all those who contributed photographs, images or assisted in the formulation of the factsheets, particularly Aarti Wadhwa (graphics) at NIWA. This project was funded by TFBIS, the Terrestrial and Freshwater Biodiversity information System (TFBIS) Programme. TFBIS is funded by the Government to help New Zealand achieve the goals of the New Zealand Biodiversity Strategy and is administered by the Department of Conservation (DOC). All photographs are by Trevor James (AgResearch), Kerry A. Bodmin or Paul D. Rushes: Champion (NIWA) unless otherwise stated. Additional images and photographs were kindly provided by Allan Herbarium; Auckland Herbarium; Larry Allain (USGS, Wetland and Aquatic Research Center); Forest and Kim Starr; Donald Cameron (Go Botany Juncus website); and Tasmanian Herbarium (Threatened Species Section, Department of Primary Industries, Parks, Water and Environment, Tasmania). factsheets © 2015 - NIWA. All rights Reserved. Cite as: Bodmin KA, Champion PD, James T & Burton T (2015) New Zealand Rushes: Juncus factsheets. NIWA, Hamilton. Introduction Rushes (family Juncaceae) are a common component of New Zealand wetland vegetation and species within this family appear very similar. With over 50 species, Juncus are the largest component of the New Zealand rushes and are notoriously difficult for amateurs and professionals alike to identify to species level. This key and accompanying factsheets have been developed to enable users with a diverse range of botanical expertise to identify Juncus to species level. The best time for collection, survey or identification is usually from December to April as mature fruiting material is required to distinguish between species. -

Fort Ord Natural Reserve Plant List
UCSC Fort Ord Natural Reserve Plants Below is the most recently updated plant list for UCSC Fort Ord Natural Reserve. * non-native taxon ? presence in question Listed Species Information: CNPS Listed - as designated by the California Rare Plant Ranks (formerly known as CNPS Lists). More information at http://www.cnps.org/cnps/rareplants/ranking.php Cal IPC Listed - an inventory that categorizes exotic and invasive plants as High, Moderate, or Limited, reflecting the level of each species' negative ecological impact in California. More information at http://www.cal-ipc.org More information about Federal and State threatened and endangered species listings can be found at https://www.fws.gov/endangered/ (US) and http://www.dfg.ca.gov/wildlife/nongame/ t_e_spp/ (CA). FAMILY NAME SCIENTIFIC NAME COMMON NAME LISTED Ferns AZOLLACEAE - Mosquito Fern American water fern, mosquito fern, Family Azolla filiculoides ? Mosquito fern, Pacific mosquitofern DENNSTAEDTIACEAE - Bracken Hairy brackenfern, Western bracken Family Pteridium aquilinum var. pubescens fern DRYOPTERIDACEAE - Shield or California wood fern, Coastal wood wood fern family Dryopteris arguta fern, Shield fern Common horsetail rush, Common horsetail, field horsetail, Field EQUISETACEAE - Horsetail Family Equisetum arvense horsetail Equisetum telmateia ssp. braunii Giant horse tail, Giant horsetail Pentagramma triangularis ssp. PTERIDACEAE - Brake Family triangularis Gold back fern Gymnosperms CUPRESSACEAE - Cypress Family Hesperocyparis macrocarpa Monterey cypress CNPS - 1B.2, Cal IPC -

Edible Seeds and Grains of California Tribes
National Plant Data Team August 2012 Edible Seeds and Grains of California Tribes and the Klamath Tribe of Oregon in the Phoebe Apperson Hearst Museum of Anthropology Collections, University of California, Berkeley August 2012 Cover photos: Left: Maidu woman harvesting tarweed seeds. Courtesy, The Field Museum, CSA1835 Right: Thick patch of elegant madia (Madia elegans) in a blue oak woodland in the Sierra foothills The U.S. Department of Agriculture (USDA) prohibits discrimination in all its pro- grams and activities on the basis of race, color, national origin, age, disability, and where applicable, sex, marital status, familial status, parental status, religion, sex- ual orientation, genetic information, political beliefs, reprisal, or because all or a part of an individual’s income is derived from any public assistance program. (Not all prohibited bases apply to all programs.) Persons with disabilities who require alternative means for communication of program information (Braille, large print, audiotape, etc.) should contact USDA’s TARGET Center at (202) 720-2600 (voice and TDD). To file a complaint of discrimination, write to USDA, Director, Office of Civil Rights, 1400 Independence Avenue, SW., Washington, DC 20250–9410, or call (800) 795-3272 (voice) or (202) 720-6382 (TDD). USDA is an equal opportunity provider and employer. Acknowledgments This report was authored by M. Kat Anderson, ethnoecologist, U.S. Department of Agriculture, Natural Resources Conservation Service (NRCS) and Jim Effenberger, Don Joley, and Deborah J. Lionakis Meyer, senior seed bota- nists, California Department of Food and Agriculture Plant Pest Diagnostics Center. Special thanks to the Phoebe Apperson Hearst Museum staff, especially Joan Knudsen, Natasha Johnson, Ira Jacknis, and Thusa Chu for approving the project, helping to locate catalogue cards, and lending us seed samples from their collections. -

Mountain Plants of Northeastern Utah
MOUNTAIN PLANTS OF NORTHEASTERN UTAH Original booklet and drawings by Berniece A. Andersen and Arthur H. Holmgren Revised May 1996 HG 506 FOREWORD In the original printing, the purpose of this manual was to serve as a guide for students, amateur botanists and anyone interested in the wildflowers of a rather limited geographic area. The intent was to depict and describe over 400 common, conspicuous or beautiful species. In this revision we have tried to maintain the intent and integrity of the original. Scientific names have been updated in accordance with changes in taxonomic thought since the time of the first printing. Some changes have been incorporated in order to make the manual more user-friendly for the beginner. The species are now organized primarily by floral color. We hope that these changes serve to enhance the enjoyment and usefulness of this long-popular manual. We would also like to thank Larry A. Rupp, Extension Horticulture Specialist, for critical review of the draft and for the cover photo. Linda Allen, Assistant Curator, Intermountain Herbarium Donna H. Falkenborg, Extension Editor Utah State University Extension is an affirmative action/equal employment opportunity employer and educational organization. We offer our programs to persons regardless of race, color, national origin, sex, religion, age or disability. Issued in furtherance of Cooperative Extension work, Acts of May 8 and June 30, 1914, in cooperation with the U.S. Department of Agriculture, Robert L. Gilliland, Vice-President and Director, Cooperative Extension -

Nymphaea Folia Naturae Bihariae Xli
https://biblioteca-digitala.ro MUZEUL ŢĂRII CRIŞURILOR NYMPHAEA FOLIA NATURAE BIHARIAE XLI Editura Muzeului Ţării Crişurilor Oradea 2014 https://biblioteca-digitala.ro 2 Orice corespondenţă se va adresa: Toute correspondence sera envoyée à l’adresse: Please send any mail to the Richten Sie bitte jedwelche following adress: Korrespondenz an die Addresse: MUZEUL ŢĂRII CRIŞURILOR RO-410464 Oradea, B-dul Dacia nr. 1-3 ROMÂNIA Redactor şef al publicațiilor M.T.C. Editor-in-chief of M.T.C. publications Prof. Univ. Dr. AUREL CHIRIAC Colegiu de redacţie Editorial board ADRIAN GAGIU ERIKA POSMOŞANU Dr. MÁRTON VENCZEL, redactor responsabil Comisia de referenţi Advisory board Prof. Dr. J. E. McPHERSON, Southern Illinois Univ. at Carbondale, USA Prof. Dr. VLAD CODREA, Universitatea Babeş-Bolyai, Cluj-Napoca Prof. Dr. MASSIMO OLMI, Universita degli Studi della Tuscia, Viterbo, Italy Dr. MIKLÓS SZEKERES Institute of Plant Biology, Szeged Lector Dr. IOAN SÎRBU Universitatea „Lucian Blaga”,Sibiu Prof. Dr. VASILE ŞOLDEA, Universitatea Oradea Prof. Univ. Dr. DAN COGÂLNICEANU, Universitatea Ovidius, Constanţa Lector Univ. Dr. IOAN GHIRA, Universitatea Babeş-Bolyai, Cluj-Napoca Prof. Univ. Dr. IOAN MĂHĂRA, Universitatea Oradea GABRIELA ANDREI, Muzeul Naţional de Ist. Naturală “Grigora Antipa”, Bucureşti Fondator Founded by Dr. SEVER DUMITRAŞCU, 1973 ISSN 0253-4649 https://biblioteca-digitala.ro 3 CUPRINS CONTENT Botanică Botany VASILE MAXIM DANCIU & DORINA GOLBAN: The Theodor Schreiber Herbarium in the Botanical Collection of the Ţării Crişurilor Museum in -

Compositae Giseke (1792)
Multequina ISSN: 0327-9375 [email protected] Instituto Argentino de Investigaciones de las Zonas Áridas Argentina VITTO, LUIS A. DEL; PETENATTI, E. M. ASTERÁCEAS DE IMPORTANCIA ECONÓMICA Y AMBIENTAL. PRIMERA PARTE. SINOPSIS MORFOLÓGICA Y TAXONÓMICA, IMPORTANCIA ECOLÓGICA Y PLANTAS DE INTERÉS INDUSTRIAL Multequina, núm. 18, 2009, pp. 87-115 Instituto Argentino de Investigaciones de las Zonas Áridas Mendoza, Argentina Disponible en: http://www.redalyc.org/articulo.oa?id=42812317008 Cómo citar el artículo Número completo Sistema de Información Científica Más información del artículo Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Página de la revista en redalyc.org Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto ISSN 0327-9375 ASTERÁCEAS DE IMPORTANCIA ECONÓMICA Y AMBIENTAL. PRIMERA PARTE. SINOPSIS MORFOLÓGICA Y TAXONÓMICA, IMPORTANCIA ECOLÓGICA Y PLANTAS DE INTERÉS INDUSTRIAL ASTERACEAE OF ECONOMIC AND ENVIRONMENTAL IMPORTANCE. FIRST PART. MORPHOLOGICAL AND TAXONOMIC SYNOPSIS, ENVIRONMENTAL IMPORTANCE AND PLANTS OF INDUSTRIAL VALUE LUIS A. DEL VITTO Y E. M. PETENATTI Herbario y Jardín Botánico UNSL, Cátedras Farmacobotánica y Famacognosia, Facultad de Química, Bioquímica y Farmacia, Universidad Nacional de San Luis, Ej. de los Andes 950, D5700HHW San Luis, Argentina. [email protected]. RESUMEN Las Asteráceas incluyen gran cantidad de especies útiles (medicinales, agrícolas, industriales, etc.). Algunas han sido domesticadas y cultivadas desde la Antigüedad y otras conforman vastas extensiones de vegetación natural, determinando la fisonomía de numerosos paisajes. Su uso etnobotánico ha ayudado a sustentar numerosos pueblos. Hoy, unos 40 géneros de Asteráceas son relevantes en alimentación humana y animal, fuentes de aceites fijos, aceites esenciales, forraje, miel y polen, edulcorantes, especias, colorantes, insecticidas, caucho, madera, leña o celulosa. -

How Does Genome Size Affect the Evolution of Pollen Tube Growth Rate, a Haploid Performance Trait?
Manuscript bioRxiv preprint doi: https://doi.org/10.1101/462663; this version postedClick April here18, 2019. to The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv aaccess/download;Manuscript;PTGR.genome.evolution.15April20 license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Effects of genome size on pollen performance 2 3 4 5 How does genome size affect the evolution of pollen tube growth rate, a haploid 6 performance trait? 7 8 9 10 11 John B. Reese1,2 and Joseph H. Williams2 12 Department of Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 13 37996, U.S.A. 14 15 16 17 1Author for correspondence: 18 John B. Reese 19 Tel: 865 974 9371 20 Email: [email protected] 21 1 bioRxiv preprint doi: https://doi.org/10.1101/462663; this version posted April 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 22 ABSTRACT 23 Premise of the Study – Male gametophytes of most seed plants deliver sperm to eggs via a 24 pollen tube. Pollen tube growth rates (PTGRs) of angiosperms are exceptionally rapid, a pattern 25 attributed to more effective haploid selection under stronger pollen competition. Paradoxically, 26 whole genome duplication (WGD) has been common in angiosperms but rare in gymnosperms.