Aki Frontiers Table of Contents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pathophysiology of Acid Base Balance: the Theory Practice Relationship
Intensive and Critical Care Nursing (2008) 24, 28—40 ORIGINAL ARTICLE Pathophysiology of acid base balance: The theory practice relationship Sharon L. Edwards ∗ Buckinghamshire Chilterns University College, Chalfont Campus, Newland Park, Gorelands Lane, Chalfont St. Giles, Buckinghamshire HP8 4AD, United Kingdom Accepted 13 May 2007 KEYWORDS Summary There are many disorders/diseases that lead to changes in acid base Acid base balance; balance. These conditions are not rare or uncommon in clinical practice, but every- Arterial blood gases; day occurrences on the ward or in critical care. Conditions such as asthma, chronic Acidosis; obstructive pulmonary disease (bronchitis or emphasaemia), diabetic ketoacidosis, Alkalosis renal disease or failure, any type of shock (sepsis, anaphylaxsis, neurogenic, cardio- genic, hypovolaemia), stress or anxiety which can lead to hyperventilation, and some drugs (sedatives, opoids) leading to reduced ventilation. In addition, some symptoms of disease can cause vomiting and diarrhoea, which effects acid base balance. It is imperative that critical care nurses are aware of changes that occur in relation to altered physiology, leading to an understanding of the changes in patients’ condition that are observed, and why the administration of some immediate therapies such as oxygen is imperative. © 2007 Elsevier Ltd. All rights reserved. Introduction the essential concepts of acid base physiology is necessary so that quick and correct diagnosis can The implications for practice with regards to be determined and appropriate treatment imple- acid base physiology are separated into respi- mented. ratory acidosis and alkalosis, metabolic acidosis The homeostatic imbalances of acid base are and alkalosis, observed in patients with differing examined as the body attempts to maintain pH bal- aetiologies. -

Increased Anion Gap Metabolic Acidosis As a Result of 5-Oxoproline (Pyroglutamic Acid): a Role for Acetaminophen
CJASN ePress. Published on April 19, 2006 as doi: 10.2215/CJN.01411005 Original Articles Increased Anion Gap Metabolic Acidosis as a Result of 5-Oxoproline (Pyroglutamic Acid): A Role for Acetaminophen Andrew Z. Fenves,* Haskell M. Kirkpatrick, III,* Viralkumar V. Patel,* Lawrence Sweetman,† and Michael Emmett* *Department of Internal Medicine, †Metabolic Disease Center, BRI Baylor University Medical Center, Dallas, Texas The endogenous organic acid metabolic acidoses that occur commonly in adults include lactic acidosis; ketoacidosis; acidosis that results from the ingestion of toxic substances such as methanol, ethylene glycol, or paraldehyde; and a component of the acidosis of kidney failure. Another rare but underdiagnosed cause of severe, high anion gap metabolic acidosis in adults is that due to accumulation of 5-oxoproline (pyroglutamic acid). Reported are four patients with this syndrome, and reviewed are 18 adult patients who were reported previously in the literature. Twenty-one patients had major exposure to acetaminophen (one only acute exposure). Eighteen (82%) of the 22 patients were women. Most of the patients were malnourished as a result of multiple medical comorbidities, and most had some degree of kidney dysfunction or overt failure. The chronic ingestion of acetaminophen, especially by malnourished women, may generate high anion gap metabolic acidosis. This undoubtedly is an underdiagnosed condition because measurements of serum and/or urinary 5-oxoproline levels are not readily available. Clin J Am Soc Nephrol 1: 441–447, 2006. doi: 10.2215/CJN.01411005 he endogenous organic acid metabolic acidoses that (3) and in patients who were taking acetaminophen (4); the occur most frequently in adults are lactic acidosis and anticonvulsant vigabatrin (5); or several antibiotics, including T ketoacidosis. -

11.3 Disorders of Acid-Base Homeostasis
674 Chapter 11. Fluids and electrolytes ( H. Sap´akov´a, D. Maasov´a) apparatus is very acidic (pH < 5.0). On the con- trary, mitochondrial compartment is slightly more 11.3 Disorders of acid-base basic than the cytosole (pH 6.7–7.2). It is difficult to measure the intracellular pH. As a consequence, homeostasis only measurements of pH of ECF(blood or plasma) are used in clinical praxis. 11.3.1.1 Sources of hydrogen ions 11.3.1 Regulation mechanisms of There are two main sources of hydrogen ions in hu- acid-base homeostasis man body: 1. the metabolism of proteins and phospholipids One of the conditions to maintain the stability of inner environment is the isohydria, i.e. the stabil- and the incomplete metabolism of fatty acids and carbohydrates. Formed acids (so called non- ity of hydrogen ion concentration in the organism. volatile acids) are no further dissociated, and Since the concentration of hydrogen ions in body flu- they must be eliminated by kidneys, ids represents a very small number (e.g. in the blood 0.00004meq/l), it is commonly expressed as pH. The 2. the complete metabolism of fatty acids and pH is defined as the negative decadic logarithm of the carbohydrates, whereby CO2 is formed. Even molar H+ concentration: pH = − log H+. The pH in though CO2 is not an acid, in the solution it is biological systems has a specific significance. The hydrated to carbonic acid which is the source of electrochemical potential of ions is proportional not + → → + − H :CO2 +H2O H2CO3 H +HCO3 . -

Lactic Acidosis Update for Critical Care Clinicians
J Am Soc Nephrol 12: S15–S19, 2001 Lactic Acidosis Update for Critical Care Clinicians FRIEDRICH C. LUFT Franz Volhard Clinic and Max Delbrück Center for Molecular Medicine, Medical Faculty of the Charité Humboldt University of Berlin, Berlin, Germany. Abstract. Lactic acidosis is a broad-anion gap metabolic aci- involves patients with underlying severe renal and cardiac dosis caused by lactic acid overproduction or underutilization. dysfunction. Drugs used to treat lactic acidosis can aggravate The quantitative dimensions of these two mechanisms com- the condition. NaHCO3 increases lactate production. Treatment monly differ by 1 order of magnitude. Overproduction of lactic of type A lactic acidosis is particularly unsatisfactory. acid, also termed type A lactic acidosis, occurs when the body NaHCO3 is of little value. Carbicarb is a mixture of Na2CO3 must regenerate ATP without oxygen (tissue hypoxia). Circu- and NaHCO3 that buffers similarly to NaHCO3 but without net latory, pulmonary, or hemoglobin transfer disorders are com- generation of CO2. The results from animal studies are prom- monly responsible. Overproduction of lactate also occurs with ising; however, clinical trials are sparse. Dichloroacetate stim- cyanide poisoning or certain malignancies. Underutilization ulates pyruvate dehydrogenase and improves laboratory val- involves removal of lactic acid by oxidation or conversion to ues, but unfortunately not survival rates, among patients with glucose. Liver disease, inhibition of gluconeogenesis, pyruvate lactic acidosis. Hemofiltration has been advocated for the dehydrogenase (thiamine) deficiency, and uncoupling of oxi- treatment of lactic acidosis, on the basis of anecdotal experi- dative phosphorylation are the most common causes. The ences. However, kinetic studies of lactate removal do not kidneys also contribute to lactate removal. -

High Anion Gap Metabolic Acidosis Due to Euglycemic Diabetic Keto
Magbri A, El-Magbri E, El-Magbri M, Brar B, Rashid S. J Cardiol and Cardiovasc Sciences (2017) 1(1): 21-23 Case Report Open Access High Anion Gap Metabolic Acidosis due to Euglycemic Diabetic Keto- acidosis Caused by Sodium-Glucose Co-transporter 2 inhibitor Awad Magbri, Eusera El-Magbri, Mariam El-Magbri, Brar Balhinder and Shauket Rashid Toledo Vascular Access Clinic, Toledo, OH, USA Article Info Case history Article Notes The case is that of 58 year-male with type 2 diabetes mellitus Received: December 05, 2017 for 7 years, hypertension, hypercholesterolemia, who was admitted Accepted: December 26, 2017 to the hospital with left lower limb cellulitis over the past 8 days. *Correspondence: On work-up he was found to have high anion-gap metabolic acidosis Dr. Awad Magbri, Toledo Vascular Access Clinic, Toledo, OH, (AGMA) with anion gap of 25, his lactate levels were normal (D USA; Email: [email protected] and L-lactate). He denies overdosing with any medications and his © 2017 Magbri A. This article is distributed under the terms of toxicology screen for methanol, ethanol, aspirin, and ethylene glycol the Creative Commons Attribution 4.0 International License. were negative. He has no psychiatric history of note. He denies using Keywords over the counter medications like acetaminophen. No bowel surgery Diabetes mellitus could be elicited. He felt dehydrated and nauseous but otherwise Diabetic ketoacidosis Euglycemic diabetic ketoacidosis Sodium-glucose cotransporter 2 inhibitor fine.His medications includes; carvedalol 25mg twice daily, Metabolic acidosis hydrochlothiazide 25 mg daily, Lipitor 20 mg daily, insulin, aspirin High anion gap acidosis ago to control his blood sugar level and A1C. -

Identifying Prognostic Factors of Severe Metabolic
Mzumara et al. Malar J (2021) 20:282 https://doi.org/10.1186/s12936-021-03785-0 Malaria Journal RESEARCH Open Access Identifying prognostic factors of severe metabolic acidosis and uraemia in African children with severe falciparum malaria: a secondary analysis of a randomized trial Grace Mzumara1,2,3,4* , Stije Leopold4,6, Kevin Marsh4,5, Arjen Dondorp4,6, Eric O. Ohuma3,4,7† and Mavuto Mukaka2,3,4,6† Abstract Background: Severe metabolic acidosis and acute kidney injury are major causes of mortality in children with severe malaria but are often underdiagnosed in low resource settings. Methods: A retrospective analysis of the ‘Artesunate versus quinine in the treatment of severe falciparum malaria in African children’ (AQUAMAT) trial was conducted to identify clinical features of severe metabolic acidosis and uraemia in 5425 children from nine African countries. Separate models were ftted for uraemia and severe metabolic acidosis. Separate univariable and multivariable logistic regression were performed to identify prognostic factors for severe metabolic acidosis and uraemia. Both analyses adjusted for the trial arm. A forward selection approach was used for model building of the logistic models and a threshold of 5% statistical signifcance was used for inclusion of variables into the fnal logistic model. Model performance was assessed through calibration, discrimination, and internal valida- tion with bootstrapping. Results: There were 2296 children identifed with severe metabolic acidosis and 1110 with uraemia. Prognostic features of severe metabolic acidosis among them were deep breathing (OR: 3.94, CI 2.51–6.2), hypoglycaemia (OR: 5.16, CI 2.74–9.75), coma (OR: 1.72 CI 1.17–2.51), respiratory distress (OR: 1.46, CI 1.02–2.1) and prostration (OR: 1.88 CI 1.35–2.59). -

Serum Bicarbonate and Dehydration Severity in Gastroenteritis 71
70 Arch Dis Child 1998;78:70–71 Serum bicarbonate and dehydration severity in Arch Dis Child: first published as 10.1136/adc.78.1.70 on 1 January 1998. Downloaded from gastroenteritis Hassib Narchi Abstract The degree of dehydration was estimated as The concentration of bicarbonate was mild, moderate, or severe by a paediatrician or measured in serum samples from 106 an emergency room doctor (table 1). Concen- children with gastroenteritis and dehy- trations of urea, creatinine, electrolytes, and dration. A concentration less than 22 bicarbonate in serum were measured on a mmol/l was more common in children venous specimen before treatment. A reduced with severe dehydration, but the magni- serum bicarbonate concentration was defined tude of bicarbonate reduction was not sig- as less than 22 mmol/l and an increased anion nificantly diVerent with increasing gap as greater than 16 mmol/l. Blood pH was degrees of dehydration. Doctors should not routinely measured. The glomerular filtra- not rely on the serum bicarbonate concen- tion rate (GFR) was estimated by the formula tration when assessing fluid deficit. devised by Schwartz et al.6 It was defined as (Arch Dis Child 1998;78:70–71) reduced if it fell two standard deviations below the mean for age and sex. The specific gravity Keywords: dehydration; gastroenteritis; serum bicarbo- and pH of urine were measured on the first nate urine sample obtained at presentation. Rehy- dration treatment (by mouth or intravenous) was prescribed at the discretion of the attend- Metabolic acidosis occurs in dehydrated pa- ing doctor. tients with gastroenteritis; there are multiple Statistical analysis of the results was carried causes of this acidosis.1–5 It is generally believed out by the Mann-Whitney test to compare two that acidosis, equated with a reduced concen- means, analysis of variance for more than two tration of bicarbonate in serum, reflects the means, the Kruskal-Wallis one way analysis of severity of dehydration, although no study sub- variance for non-parametric means, and the 2 stantiating this has been found. -

ACS/ASE Medical Student Core Curriculum Acid-Base Balance
ACS/ASE Medical Student Core Curriculum Acid-Base Balance ACID-BASE BALANCE Epidemiology/Pathophysiology Understanding the physiology of acid-base homeostasis is important to the surgeon. The two acid-base buffer systems in the human body are the metabolic system (kidneys) and the respiratory system (lungs). The simultaneous equilibrium reactions that take place to maintain normal acid-base balance are: H" HCO* ↔ H CO ↔ H O l CO g To classify the type of disturbance, a blood gas (preferably arterial) and basic metabolic panel must be obtained. A basic understanding of normal acid-base buffer physiology is required to understand alterations in these labs. The normal pH of human blood is 7.40 (7.35-7.45). This number is tightly regulated by the two buffer systems mentioned above. The lungs contain carbonic anhydrase which is capable of converting carbonic acid to water and CO2. The respiratory response results in an alteration to ventilation which allows acid to be retained or expelled as CO2. Therefore, bradypnea will result in respiratory acidosis while tachypnea will result in respiratory alkalosis. The respiratory buffer system is fast acting, resulting in respiratory compensation within 30 minutes and taking approximately 12 to 24 hours to reach equilibrium. The renal metabolic response results in alterations in bicarbonate excretion. This system is more time consuming and can typically takes at least three to five days to reach equilibrium. Five primary classifications of acid-base imbalance: • Metabolic acidosis • Metabolic alkalosis • Respiratory acidosis • Respiratory alkalosis • Mixed acid-base disturbance It is important to remember that more than one of the above processes can be present in a patient at any given time. -

Plasmodium Falciparum Var Gene/ Important Role of the EPCR-Activated Protein C (APC) Pathway in P
Severe adult malaria is associated with specific PNAS PLUS PfEMP1 adhesion types and high parasite biomass Maria Bernabeua, Samuel A. Danzigera, Marion Avrila, Marina Vazb, Prasad H. Babarc,d, Andrew J. Braziera, Thurston Herricksc,d, Jennifer N. Makic,d, Ligia Pereirac,d, Anjali Mascarenhasc,d, Edwin Gomesb, Laura Cheryc,d, John D. Aitchisona, Pradipsinh K. Rathodc,d, and Joseph D. Smitha,1 aCenter for Infectious Disease Research, Seattle, WA 98109; bDepartment of Medicine, Goa Medical College & Hospital, Bambolim, Goa 403202, India; cDepartment of Chemistry, University of Washington, Seattle, WA 98195; and dDepartment of Global Health, University of Washington, Seattle, WA 98195 Edited by Mats Wahlgren, Karolinska Institutet, Stockholm, Sweden, and accepted by the Editorial Board April 7, 2016 (received for review December 10, 2015) The interplay between cellular and molecular determinants that This subset of PfEMP1s includes mediators of distinct infected lead to severe malaria in adults is unexplored. Here, we analyzed erythrocyte adhesion categories, including “rosetting” and endo- parasite virulence factors in an infected adult population in India thelial protein C receptor (EPCR) binding. Rosetting involves ad- and investigated whether severe malaria isolates impair endothe- hesion to uninfected red blood cells (20, 21), possibly leading to lial protein C receptor (EPCR), a protein involved in coagulation greater microvascular obstruction (22). EPCR binding involves in- and endothelial barrier permeability. Severe malaria isolates overex- fected erythrocyte adhesion to vascular endothelial cells (23). The pressed specific members of the Plasmodium falciparum var gene/ important role of the EPCR-activated protein C (APC) pathway in P. falciparum PfEMP1 ( erythrocyte membrane protein 1) family that regulating coagulation, inflammation, and endothelial barrier prop- var bind EPCR, including DC8 genes that have previously been linked erties (24) has led to the hypothesis that EPCR-binding parasites to severe pediatric malaria. -

Metabolic Acidosis in Childhood: Why, When and How to Treat Acidose Metabólica Na Infância: Por Que, Quando E Como Tratá-La?
0021-7557/07/83-02-Suppl/S11 Jornal de Pediatria Copyright © 2007 by Sociedade Brasileira de Pediatria ARTIGO DE REVISÃO Metabolic acidosis in childhood: why, when and how to treat Acidose metabólica na infância: por que, quando e como tratá-la? Olberes V. B. Andrade1, Flávio O. Ihara2, Eduardo J. Troster3 Resumo Abstract Objetivo: Apresentar uma revisão atualizada e crítica sobre os Objectives: To critically discuss the treatment of metabolic acidosis and mecanismos das principais patologias associadaseotratamento da acidose the main mechanisms of disease associated with this disorder; and to metabólica, discutindo aspectos controversos quanto aos benefícios e riscos describe controversial aspects related to the risks and benefits of using da utilização do bicarbonato de sódio e outras formas de terapia. sodium bicarbonate and other therapies. Fontes dos dados: Revisão da literatura publicada, obtida através de Sources: Review of PubMed/MEDLINE, LILACS and Cochrane Library busca eletrônica com as palavras-chave acidose metabólica, acidose láctica, databases for articles published between 1996 and 2006 using the following cetoacidose diabética, ressuscitação cardiopulmonar, bicarbonato de sódio e keywords: metabolic acidosis, lactic acidosis, ketoacidosis, diabetic terapêutica nas bases de dados PubMed/MEDLINE, LILACS e Cochrane ketoacidosis, cardiopulmonary resuscitation, sodium bicarbonate, Library, entre 1996 e 2006, além de publicações clássicas referentes ao treatment. Classical publications concerning the topic were also reviewed. -
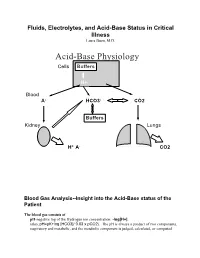
Acid-Base Physiology Cells Buffers
Fluids, Electrolytes, and Acid-Base Status in Critical Illness Laura Ibsen, M.D. Acid-Base Physiology Cells Buffers H+ Blood A- HCO3- CO2 Buffers Kidney Lungs H+ A- CO2 Blood Gas Analysis--Insight into the Acid-Base status of the Patient The blood gas consists of pH-negative log of the Hydrogen ion concentration: -log[H+]. (also, pH=pK+log [HCO3]/ 0.03 x pCO2). The pH is always a product of two components, respiratory and metabolic, and the metabolic component is judged, calculated, or computed by allowing for the effect of the pCO2, ie, any change in the pH unexplained by the pCO2 indicates a metabolic abnormality. - + CO2+H20º H2CO3ºHCO3 + H CO2 and water form carbonic acid or H2CO3, which is in equilibrium with bicarbonate (HCO3-)and hydrogen ions (H+). A change in the concentration of the reactants on either side of the equation affects the subsequent direction of the reaction. For example, an increase in CO2 will result in increased carbonic acid formation (H2CO3) which leads to an increase in both HCO3- and H+ (\pH). Normally, at pH 7.4, a ratio of one part carbonic acid to twenty parts bicarbonate is present in the extracellular fluid [HCO3-/H2CO3]=20. A change in the ratio will affect the pH of the fluid. If both components change (ie, with chronic compensation), the pH may be normal, but the other components will not. pCO2-partial pressure of carbon dioxide. Hypoventilation or hyperventilation (ie, minute ventilation--tidal volume x respitatory rate--imperfectly matched to physiologic demands) will lead to elevation or depression, respectively, in the pCO2. -

Diagnosis and Management of Metabolic Acidosis: Guidelines From
Jung et al. Ann. Intensive Care (2019) 9:92 https://doi.org/10.1186/s13613-019-0563-2 REVIEW Open Access Diagnosis and management of metabolic acidosis: guidelines from a French expert panel Boris Jung1,2*, Mikaël Martinez3,4, Yann‑Erick Claessens5, Michaël Darmon6,7,8, Kada Klouche2,9, Alexandre Lautrette10,11, Jacques Levraut12,13, Eric Maury14,15,16, Mathieu Oberlin17, Nicolas Terzi18,19, Damien Viglino20,21, Youri Yordanov22,23,24, Pierre‑Géraud Claret25, Naïke Bigé14 , for the Société de Réanimation de Langue Française (SRLF) and the Société Française de Médecine d’Urgence (SFMU) Abstract Metabolic acidosis is a disorder frequently encountered in emergency medicine and intensive care medicine. As litera‑ ture has been enriched with new data concerning the management of metabolic acidosis, the French Intensive Care Society (Société de Réanimation de Langue Française [SRLF]) and the French Emergency Medicine Society (Société Française de Médecine d’Urgence [SFMU]) have developed formalized recommendations from experts using the GRADE methodology. The felds of diagnostic strategy, patient assessment, and referral and therapeutic management were addressed and 29 recommendations were made: 4 recommendations were strong (Grade 1), 10 were weak (Grade 2), and 15 were experts’ opinions. A strong agreement from voting participants was obtained for all recom‑ mendations. The application of Henderson–Hasselbalch and Stewart methods for the diagnosis of the metabolic acidosis mechanism is discussed and a diagnostic algorithm is proposed. The use of ketosis and venous and capillary lactatemia is also treated. The value of pH, lactatemia, and its kinetics for the referral of patients in pre‑hospital and emergency departments is considered.