From Laboratory to Patient: the Journey of a Centrally Authorised Medicine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Origin of Bimaristans (Hospitals) in Islamic Medical History
The Origin of Bimaristans (Hospitals) in Islamic Medical History IMPORTANT NOTICE: Author: Dr. Sharif Kaf Al-Ghazal Chief Editor: Prof. Mohamed El-Gomati All rights, including copyright, in the content of this document are owned or controlled for these purposes by FSTC Limited. In Deputy Editor: Prof. Mohammed Abattouy accessing these web pages, you agree that you may only download the content for your own personal non-commercial Associate Editor: Dr. Salim Ayduz use. You are not permitted to copy, broadcast, download, store (in any medium), transmit, show or play in public, adapt or Release Date: April 2007 change in any way the content of this document for any other purpose whatsoever without the prior written permission of FSTC Publication ID: 682 Limited. Material may not be copied, reproduced, republished, Copyright: © FSTC Limited, 2007 downloaded, posted, broadcast or transmitted in any way except for your own personal non-commercial home use. Any other use requires the prior written permission of FSTC Limited. You agree not to adapt, alter or create a derivative work from any of the material contained in this document or use it for any other purpose other than for your personal non-commercial use. FSTC Limited has taken all reasonable care to ensure that pages published in this document and on the MuslimHeritage.com Web Site were accurate at the time of publication or last modification. Web sites are by nature experimental or constantly changing. Hence information published may be for test purposes only, may be out of date, or may be the personal opinion of the author. -

HOSPITAL BEDS and RELATED EQUIPMENT DME101.001 Bluereview POSTED DATE: 11/17/2003 EFFECTIVE DATE: 2/27/2004 ______
HOSPITAL BEDS AND RELATED EQUIPMENT DME101.001 BlueReview POSTED DATE: 11/17/2003 EFFECTIVE DATE: 2/27/2004 _____________________________________________________________________________ COVERAGE: HOSPITAL BEDS Various types of hospital beds may be medically necessary when the selected appropriate criteria are met: A. FIXED HEIGHT (one or more of the following is required): · The patient requires positioning of the body for the alleviation of pain in ways not feasible with an ordinary bed. · The patient requires the head of the bed to be elevated more than 30 degrees most of the time due to congestive heart failure, chronic pulmonary disease, or problems with aspiration. Pillows or wedges should first have been considered. · The patient requires traction equipment that can only be attached to a hospital bed. B. VARIABLE HEIGHT (in addition to one of the fixed height criteria): · The patient requires a bed height different than a fixed height hospital bed to permit transfers to chair, wheelchair or standing position. C. SEMI-ELECTRIC: · In addition to the all indications for A and B, the patient requires frequent or immediate changes in body position. D. TOTAL ELECTRIC: · This type of bed is rarely indicated EXCEPT in cases of spinal cord injuries, brain injury patients, and patients with neurological damage that prevents them from getting in or out of bed. These patients also require assistance with the basic activities of daily living (i.e., bathing, use of toilet). E. HEAVY DUTY EXTRA WIDE: · Is covered if the patient meets one of the criteria for a fixed height hospital bed and patient weight is more than 350 pounds but does not exceed 600 pounds. -

Orderly / Patient Attendant Role Purpose
ORDERLY / PATIENT ATTENDANT Four shifts in a 2-week period, plus availability (6:00am – 2:00pm, 2:00pm – 10:00pm) Reference #: PAB0002 We are the largest free-standing palliative care residence in Canada with 23 beds, serving the needs of our community. We offer quality end of life care for patients and their families. We are looking for someone to join our team to fill the position of Orderly / Patient Attendant to work 4 shifts in a 2-week period plus availability. ROLE PURPOSE: Reporting to the Nurse Manager, the Orderly / Patient Attendant works within the interdisciplinary team of nursing and volunteers to provide safe comfort care and assistance to patients for activities of daily living (under the supervision or assistance of an RN or LPN). PRIMARY RESPONSIBILITIES: • Provides hygienic care to the patient using means necessary to maintain patient safety and comfort • Gives routine mouth, skin care; shaves the patient as needed • Makes beds - occupied and unoccupied • Immediately reports to a nurse any changes in the patient’s condition • With thoughtfulness, by skillfully avoiding discomfort to the patient, employs gentle turning and positioning techniques • Safely lifts and/or transfers patients using proper body mechanics • Assists patients to ambulate safely • Feeds patients, or offers assistance in feeding (set up, positioning, etc.) • Uses proper techniques to feed patients at risk for aspiration due to their medical condition • Assists patients in the safe use of bedpan, urinal, bedside commode or assists patients to/from the bathroom • Empties urinary drainage bags and notes the amount, color & consistency to the nurse • Assists in activities related to the death of a patient (i.e. -

Orderly, Medical Services
International Hazard Datasheets on Occupation Orderly, medical services What is a Hazard Datasheet on Occupation? This datasheet is one of the International Datasheets on Occupations. It is intended for those professionally concerned with health and safety at work: occupational physicians and nurses, safety engineers, hygienists, education and Information specialists, inspectors, employers ' representatives, workers' representatives, safety officers and other competent persons. This datasheet lists, in a standard format, different hazards to which orderly, medical servicess may be exposed in the course of their normal work. This datasheet is a source of information rather than advice. With the knowledge of what causes injuries and diseases, is easier to design and implement suitable measures towards prevention. This datasheet consists of four pages: Page 1: Information on the most relevant hazards related to the occupation. Page 2: A more detailed and systematized presentation on the different hazards related to the job with indicators for preventive measures (marked as and explained on the third page). Page 3: Suggestions for preventive measures for selected hazards. Page 4: Specialized information, relevant primarily to occupational safety and health professionals and including information such as a brief job description, a list of tasks, notes and references. Who is an orderly? A healthcare worker who performs various tasks as directed by nurses and other medical staff. Responsible for feeding, bathing, and massaging patients, and also for transfer of hospitalized patients from one ward to another. What is dangerous about this job? Orderlies are exposed to practically all of the acute hazards existing in the healthcare institutions where they work. Orderlies may be exposed to infectious diseases due to direct contact with patients and their body fluids. -

The Impact of Bimaristans Design on Design Factors of Therapeutic Buildings an Environmental Field Study in Makkah, Saudi Arabia
59 Al Anoud Al Ansary & Hirao Kazuhiro The Impact of Bimaristans Design on Design Factors of Therapeutic Buildings An Environmental Field Study in Makkah, Saudi Arabia Alanoud Alansari, Contact Author “[email protected]” Hirao Kazuhiro KAZUHIRO HIRAO, Department of Architecture and Urban Design, Ritsumeikan University, Japan Abstract: Keywords: "Paradise Garden" is a concept used in Bimaristans "Islamic historical therapeutic Therapeutic buildings buildings" which has given rise to the application of several spiritual and aesthetic Paradise Principle meanings that helped to raise the efficiency of the therapeutic environment. The Bimaristan hypothesis revolves around the idea that the technological development increases the interest in the functional side rather than the aesthetic side in the current therapeutic buildings. This had a negative impact on the efficiency of the functional environment which exceeded to the satisfaction and comfort of the buildings’ users. This hypothesis has been tested through two types of studies: The first study was an ‘Analytical Study’ (of 4 hospitals) analyzing the horizontal projections to measure the effects of the natural lighting and ventilation. In the second study, questionnaires were distributed to both patients and staff in the therapeutic buildings to measure the therapeutic environment efficiency and the extent of satisfaction among of the buildings’ users. It was concluded that the natural lighting and ventilation drive up healing (treatment) rate in the therapeutic environment. This concluded that the technological advances in the medical field helped to raise the level of functional performance and thus replaced a large part of the role of the natural lighting and ventilation. Paper received 17th November 2016, Accepted 26th November 2016, Published 15st of January 2017 1. -

Update on Hospital Medicine ACP TN Scientific Meeting 2019
Update on Hospital Medicine ACP TN Scientific Meeting 2019 Chase J. Webber, DO Assistant Professor Clinical Medicine Vanderbilt University Medical Center Section of Hospital Medicine @chasejwebber COI • I have no actual or potential conflict of interest in relation to this presentation. Thinking about Hospital Medicine Thinking about Hospital Medicine Where we are, where we’re going, we’re we’ve been Courtesy: Tennessee State Library and Archives 1994 “Before the Titans, TV shows and pedal taverns” 2019 N Engl J Med 2016; 375:1009-1011 Growth in the Number of Hospitalists in the United States, 2003–2016. N Engl J Med 2016; 375:1009-1011 Old standards. New hits… Timeless Pearls! 1 Best Practices in Medication Reconciliation 2 Antibiotic Stewardship 2018- 2019 3 Delirium management Updates 4 Discharge AMA 5 Discharge Pearls*** Best Practices – Medication Reconciliation CHAOS MEDICATION TRANSITION ADEs Random Common Systematic?? Gold chaos sense Med Rec Standard Med Rec Clinical question – Which Med Rec intervention is most effective at reducing inpatient medication discrepancies? Study design – Mentored, Quality Improvement study Setting – 791 patients in 5 hospitals over 25 months J Hosp Medicine, epub: 8/21/19 DOI 10.12788/jhm.3308 Interventions and Results J Hosp Medicine, epub: 8/21/19 DOI 10.12788/jhm.3308 What stands out? ?Potential for ADEs ?Omitted medications Sources: need at least 2 ?Handwritten vs. EMR Interdisciplinary input Diverse mix -carried over -OTC -vitamins -supplements -nonhelpful or harmful • QR code to access Marquis/SHM resources portal Timeless Pearls • Seek to obtain a Best Possible Medication History (BPMH) on admission. • Specially trained Pharmacy staff and support: essential. -

Admission of Patients APPROVAL SIGNATURE
CASCADE VALLEY HOSPITAL AND CLINICS POLICY & PROCEDURE NUMBER: 53-3-001 DATE EFFECTIVE: 12/90 TITLE: Admission of Patients APPROVAL SIGNATURE: REVIEW REVISE DATE DATE SIGNATURE 12/04 DIVISION: CVH DEPARTMENT: Registration 12/07 12/07 ROUTE TO: Registration 12/10 12/10 12/13 CROSS REFERENCE: 53-3-03, 53-3-46 6/14 6/14 SCOPE This procedure applies to all employees whose terms and conditions of employment are administered by Cascade Valley Hospital and Clinics. PROCEDURE All patients receiving services at CVH will be registered using one of the following admitting functions: INPATIENT (IN) A patient who is assigned a room and bed and receives automatic room/bed charges. OBSERVATION (INO) An outpatient who is assigned a room and bed, but does not receive an automatic room/bed charge. RECURRING (RCR) An outpatient who returns to the hospital more than two times for related treatments (a series patient). Examples: Oncology, Recurring meds. Initial visit of the month is assigned a new account number, subsequent visits are entered under "revisit" function. The same account number is used for an entire month. See also 53-3-03. REFERRED (REF) Used to register CLIENTS (See Procedure 53-3-46) and Diabetes Education MNT patients. EMERGENCY (ER) An outpatient who comes to the hospital unscheduled and is treated in the emergency room. SURGICAL DAY CARE (SDC) A patient who visits the hospital for outpatient surgery. CLINICAL (CLI) Used for outpatient visits (Lab, Radiology, Respiratory Therapy, Same Day Procedure, NST, OB Check). ||NEWBORN ||A newborn baby patient who is assigned a nursery room bed and ||receives automatic nursery charges. -

Hospital Medicine and Hospitalists
JAMA PATIENT PAGE | Health Care Delivery Hospital Medicine and Hospitalists Hospital medicine is a field of medicine that is dedicated to providing patients with high-quality care during their hospital stay. Hospital medicine is the fastest-growing medical specialty in the theyarefirstadmittedtothehospital.Hospitalistshavetogothrough United States. Doctors who practice hospital medicine are called a detailed history with their patients, which can be time consum- hospitalists. Hospitalists are usually trained in internal medicine, ing. But hospitalists will often talk to a patient’s PCP for back- pediatrics, or family medicine. ground information. Another potential downside is follow-up after discharge from the hospital. If a patient’s PCP is not involved dur- The Era of Hospital Medicine ing the patient’s hospital stay, the PCP can find out what happened In the past, when a patient was admitted to the hospital, that pa- only through medical records or the patient’s own account, both of tient’s primary care physician (PCP) would also take care of him or which are not as ideal as being present firsthand. her every day in the hospital. However, it was often difficult for PCPs The bottom line is that there should always be an open line of to see both their regularly scheduled clinic patients as well as their communication between a patient’s PCP and hospitalist. hospitalized patients every day. As a result, hospitals started hiring hospitalists, doctors who take care of patients only while they are What You Can Do as a Patient in the hospital. These doctors do not see patients in clinics or out- If you are admitted to the hospital under the care of a hospitalist, patient offices. -

Ch6 the Patient's Or Resident's Environment Q&A
Chapter 6 - Test Name: The Patient’s or Resident’s Date: Environment 1. What special safety modification might be found in a patient's or resident's bathroom? A) A call light B) A television C) A low toilet seat D) A privacy curtain 2. A patient's or resident's room is first considered his or her home when his or her A) move to the facility is permanent. B) room has a full private bath. C) room is not shared with anyone. D) admission is complete. 3. Which of the following regulations states that a resident's unit in a long-term care facility must be clean, safe, orderly, and free of obstacles in the pathway? A) Occupational Safety and Health Administration (OSHA) regulations B) Omnibus Budget Reconciliation Act (OBRA) regulations C) Medicare regulations D) Medicaid regulations 4. Who is responsible for keeping the health care facility clean? A) The nursing assistant B) The housekeeping staff C) Patients and residents D) Each member of the health care team 5. Which one of the following is the nursing assistant's regular responsibility? A) Routinely mopping the floors B) Regularly emptying the trash cans C) Laundering the facility's bed linens D) Helping residents attend to their belongings 6. Nursing assistants help to control odors in the health care facility by A) drawing the privacy curtain when a person is using the bedpan. B) emptying and cleaning used urinals promptly. C) opening the windows to air the facility out. D) changing the bed linens frequently. Page 1 7. When is Trendelenburg's position used? A) When a person's head needs to be higher than his or her feet B) When a person's head needs to be lower than his or her feet C) When a bed needs to be moved D) When a bed needs to be raised 8. -

The Key Principles and Characteristics of an Effective
The Key Principles and Characteristics of an Effective HospitalThe Medicine Key Group Principles (HMG) and Characteristics of an Effective Hospital Medicine Group: An Assessment Guide for Hospitals and Hospitalists Updated Edition Satisfaction Engaged Hospitalists Adequate Resources Quality, Safety and Efficiency Effective Leadership Management Infrastructure 1 The Key Key Principles Principles and Characteristics and Characteristics of an Effective of anHospital Effective Medicine GroupHospital (HMG) Medicine Group (HMG) Contributors Patrick J. Cawley, MD, MBA John Nelson, MD, MHM Chief Executive Officer Overlake Medical Center Medical University of South Carolina Hospitalist Practice Charleston, South Carolina Nelson Flores Hospital Medicine Consultants Steven Deitelzweig, MD Bellevue, Washington System Department Chair of Hospital Medicine Scott Rissmiller, MD Ochsner Health System President, Carolinas Hospitalist Group New Orleans, Louisiana Chief Medical Director, Adult Acute Care Carolinas Healthcare System Leslie A. Flores, MHA, SFHM Nelson Flores Hospital Medicine Laurence Wellikson, MD, SFHM Consultants Chief Executive Officer La Quinta, California Society of Hospital Medicine Philadelphia, Pennsylvania Joseph A. Miller, MS Senior Vice President Winthrop F. Whitcomb, MD, MHM Society of Hospital Medicine Chief Medical Officer Philadelphia, Pennsylvania Remedy Partners Darien, Connecticut Copyright ©2015 by Society of Hospital Medicine. All rights reserved. No part of this publication may be reproduced, stored in retrieval system, -

Pdf (Accessed 2020 June 15)
Research Impact of an internal medicine nocturnist service on care of patients with cancer at a large Canadian teaching hospital: a quality-improvement study Richard Dunbar-Yaffe MDCM MSc, Robert C. Wu MD MSc, Amit Oza MBBS, Victoria Lee-Kim BMSc, Peter Cram MD MBA Abstract Background: Nocturnists (overnight hospitalists) are commonly implemented in US teaching hospitals to adhere to per-resident patient caps and improve care but are rare in Canada, where patient caps and duty hours are comparatively flexible. Our objective was to assess the impact of a newly implemented nocturnist program on perceived quality of care, code status documentation and patient outcomes. Methods: Nocturnists were phased in between June 2018 and December 2019 at Toronto General Hospital, a large academic teaching hospital in Toronto, Ontario. We performed a quality-improvement study comparing rates of code status entry into the elec- tronic health record at admission, in-hospital mortality, the 30-day readmission rate and hospital length of stay for patients with cancer admitted by nocturnists and by residents. Surveys were administered in June 2019 to general internal medicine faculty and residents to assess their perceptions of the impact of the nocturnist program. Results: From July 2018 to June 2019, 30 nocturnists were on duty for 241/364 nights (66.5%), reducing the mean maximum over- night per-resident patient census from 40 (standard deviation [SD] 4) to 25 (SD 5) (p < 0.001). The rate of admission code sta- tus entry was 35.3% among patients admitted by residents (n = 133) and 54.9% among those admitted by nocturnists (n = 339) (p < 0.001). -
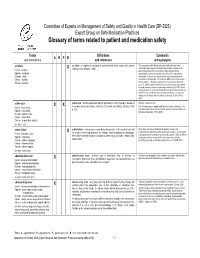
Glossary of Terms Related to Patient and Medication Safety
Committee of Experts on Management of Safety and Quality in Health Care (SP-SQS) Expert Group on Safe Medication Practices Glossary of terms related to patient and medication safety Terms Definitions Comments A R P B and translations and references and synonyms accident accident : an unplanned, unexpected, and undesired event, usually with adverse “For many years safety officials and public health authorities have Xconsequences (Senders, 1994). discouraged use of the word "accident" when it refers to injuries or the French : accident events that produce them. An accident is often understood to be Spanish : accidente unpredictable -a chance occurrence or an "act of God"- and therefore German : Unfall unavoidable. However, most injuries and their precipitating events are Italiano : incidente predictable and preventable. That is why the BMJ has decided to ban the Slovene : nesreča word accident. (…) Purging a common term from our lexicon will not be easy. "Accident" remains entrenched in lay and medical discourse and will no doubt continue to appear in manuscripts submitted to the BMJ. We are asking our editors to be vigilant in detecting and rejecting inappropriate use of the "A" word, and we trust that our readers will keep us on our toes by alerting us to instances when "accidents" slip through.” (Davis & Pless, 2001) active error X X active error : an error associated with the performance of the ‘front-line’ operator of Synonym : sharp-end error French : erreur active a complex system and whose effects are felt almost immediately. (Reason, 1990, This definition has been slightly modified by the Institute of Medicine : “an p.173) error that occurs at the level of the frontline operator and whose effects are Spanish : error activo felt almost immediately.” (Kohn, 2000) German : aktiver Fehler Italiano : errore attivo Slovene : neposredna napaka see also : error active failure active failures : actions or processes during the provision of direct patient care that Since failure is a term not defined in the glossary, its use is not X recommended.