Molecular Analysis of the Eggshell and Cuticle Surface of the Potato Cyst Nematode, Globodera Rostochiensis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pest Management Strategic Plan for Organic Potato Production in the West
Pest Management Strategic Plan for Organic Potato Production in the West Summary of workshops held on February 16, 2006 Buhl, Idaho and January 9, 2008 Portland, Oregon Issue Date December 19, 2008 Lead Authors: Jennifer Miller, Ronda Hirnyck, Lisa Downey-Blecker Editor: Diane Clarke This project was sponsored by the Western Integrated Pest Management Center, which is funded by the United States Department of Agriculture, Cooperative State Research, Education, and Extension Service. Additional funding was provided by the Organic Farming Research Foundation and the Bullitt Foundation. Table of Contents Work Group .........................................................................................................................3 Summary of the Most Critical Needs in Organic Potato Production in the West ................5 Introduction ..........................................................................................................................6 Production Overview .........................................................................................................11 California ...............................................................................................................11 Colorado .................................................................................................................12 Columbia Basin ......................................................................................................12 Idaho ......................................................................................................................13 -

PCN Guidelines, and Potato Cyst Nematodes (Globodera Rostochiensis Or Globodera Pallida) Were Not Detected.”
Canada and United States Guidelines on Surveillance and Phytosanitary Actions for the Potato Cyst Nematodes Globodera rostochiensis and Globodera pallida 7 May 2014 Table of Contents 1. Introduction ...........................................................................................................................................................3 2. Rationale for phytosanitary actions ........................................................................................................................3 3. Soil sampling and laboratory analysis procedures .................................................................................................4 4. Phytosanitary measures ........................................................................................................................................4 5. Regulated articles .................................................................................................................................................5 6. National PCN detection survey..............................................................................................................................6 7. Pest-free places of production or pest-free production sites within regulated areas ...............................................6 8. Phytosanitary certification of seed potatoes ..........................................................................................................7 9. Releasing land from regulatory control ..................................................................................................................8 -

A Transcriptomic Snapshot of Early Molecular Communication Between Pasteuria Penetrans and Meloidogyne Incognita Victor Phani1, Vishal S
Phani et al. BMC Genomics (2018) 19:850 https://doi.org/10.1186/s12864-018-5230-8 RESEARCH ARTICLE Open Access A transcriptomic snapshot of early molecular communication between Pasteuria penetrans and Meloidogyne incognita Victor Phani1, Vishal S. Somvanshi1, Rohit N. Shukla2, Keith G. Davies3,4* and Uma Rao1* Abstract Background: Southern root-knot nematode Meloidogyne incognita (Kofoid and White, 1919), Chitwood, 1949 is a key pest of agricultural crops. Pasteuria penetrans is a hyperparasitic bacterium capable of suppressing the nematode reproduction, and represents a typical coevolved pathogen-hyperparasite system. Attachment of Pasteuria endospores to the cuticle of second-stage nematode juveniles is the first and pivotal step in the bacterial infection. RNA-Seq was used to understand the early transcriptional response of the root-knot nematode at 8 h post Pasteuria endospore attachment. Results: A total of 52,485 transcripts were assembled from the high quality (HQ) reads, out of which 582 transcripts were found differentially expressed in the Pasteuria endospore encumbered J2 s, of which 229 were up-regulated and 353 were down-regulated. Pasteuria infection caused a suppression of the protein synthesis machinery of the nematode. Several of the differentially expressed transcripts were putatively involved in nematode innate immunity, signaling, stress responses, endospore attachment process and post-attachment behavioral modification of the juveniles. The expression profiles of fifteen selected transcripts were validated to be true by the qRT PCR. RNAi based silencing of transcripts coding for fructose bisphosphate aldolase and glucosyl transferase caused a reduction in endospore attachment as compared to the controls, whereas, silencing of aspartic protease and ubiquitin coding transcripts resulted in higher incidence of endospore attachment on the nematode cuticle. -
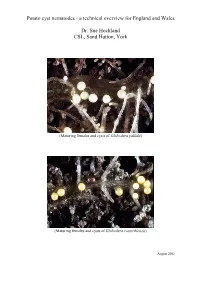
Potato Cyst Nematodes - a Technical Overview for England and Wales
Potato cyst nematodes - a technical overview for England and Wales Dr. Sue Hockland CSL, Sand Hutton, York (Maturing females and cysts of Globodera pallida) (Maturing females and cysts of Globodera rostochiensis) August 2002 Contents Page • Executive Summary 1 • Introduction 2 • PCN: species and diagnosis 2 • Biology of PCN species 3 • Detection 4 • Host plants of PCN 4 • Symptoms 5 • Damage 5 • Pathotypes and Host Plant Resistance 5 • Distribution and spread in England and Wales 6 • Distribution in Europe and elsewhere 9 • Statutory Management of PCN 11 • Management of PCN in ware potatoes 13 • Chemical methods 14 • Non-chemical methods 14 • Conclusion 16 • Acknowledgements 17 Executive Summary The pests Potato cyst nematode (PCN) is the name commonly given to two species of cyst nematode that attack potato, namely Globodera pallida (Stone) Behrens and G. rostochiensis (Wollenweber) Behrens. They are two of the most important pests of potato in England and Wales, feeding on potato roots, causing losses of yield and costs that vary and are difficult to estimate. Published papers usually quote losses of about 9% of annual yield, estimated at about £43 million for the UK, based on the mean value of the crop from 1990-1995. Adaptations to a plant-parasitic life In both species the female forms a hard covering around her eggs when she dies, creating a ‘cyst’ which protects the eggs and developing juveniles from desiccation, predation and chemical control. Only a proportion of the eggs hatch from the cyst each year, and in G. pallida this occurs at a slower rate and with a later annual peak of hatching than G. -

Pasteuria Nishizawae – Pn1 (016455) Fact Sheet
Pasteuria nishizawae – Pn1 (016455) Fact Sheet Summary Pasteuria, a genus of bacteria, includes several species that have shown potential in controlling plant- parasitic nematodes that attack and cause significant damage to many valuable agricultural crops. These gram-positive, mycelial, endospore-forming bacteria are mainly obligate parasites (i.e., organisms that depend on particular hosts to complete their own life cycle) of nematodes, although one species, Pasteuria ramosa, is known to parasitize water fleas. Pasteuria species are ubiquitous in most environments and are found in nematodes in at least 80 countries on 5 continents, as well as on islands in the Atlantic, Pacific, and Indian Oceans. In light of the demonstrated nematicidal capabilities and host specificity of Pasteuria nishizawae – Pn1, Pasteuria Bioscience, Inc. proposed to register a manufacturing-use pesticide product, Soyacyst Tech, and two end-use pesticide products, Soyacyst Tech+ and Soyacyst LF, containing this bacterium. Soyacyst Tech+ and Soyacyst LF will be applied to soybean or its seed to control the soybean cyst nematode. Use of Pasteuria nishizawae – Pn1 as a nematicide and in accordance with label directions is not expected to cause any unreasonable adverse effects on human health or the environment. I. Description of the Active Ingredient Pasteuria nishizawae – Pn1 was isolated from an Illinois soybean field in the mid-2000s. Although endospores of Pasteuria nishizawae have been observed to attach to the cuticle of three nematodes of the genus Heterodera and one nematode of the genus Globodera, it is known only to infect and complete its life cycle within the female soybean cyst nematode (Heterodera glycines). -

A Mathematic Model for the Interaction Between Meloidogyne Spp. and Pasteuria Penetrans
Rev. Protección Veg. Vol. 29 No. 2 (2014): 145-149 TECHNICAL NOTE A mathematic model for the interaction between Meloidogyne spp. and Pasteuria penetrans Ileana MirandaI, Lucila GómezI, Hugo L. BenítezII, Yoannia CastilloII, Dainé Hernández-OchandíaI, Mayra G. RodríguezI I Dirección Sanidad Vegetal y II Dirección de Ciencia, Innovación y Postgrado. Centro Nacional de Sanidad Agropecuaria (CENSA), Apartado 10, San José de las Lajas, Mayabeque, Cuba. Email: [email protected]. ABSTRACT: Some aspects of Meloidogyne spp. (root-knot nematode) growth regulation by the gram- negative bacterium Pasteuria penetrans were reviewed. The study allowed the construction of a mathematical model to predict the dynamics of both organisms. The model includes 11 differential equations and 31biological constants describing the life-cycle of the nematode and its relationship with the bacterium. To simulate the real behavior of the populations, the constants, which represent biological parameters, have to be evaluated under controlled conditions similar to those of soil microcosm. Key words: bio-mathematics, Meloidogyne, Pasteuria penetrans, biological control. Modelo matemáticode la interacción entre Meloidogyne spp. y Pasteuria penetrans RESUMEN: El estudio de algunos aspectos de la regulación del crecimiento de los nematodos agalleros del género Meloidogyne Goeldi por la acción de la bacteria Pasteuria penetrans, permitió proponer un modelo matemático para predecir la dinámica de ambos organismos. El modelo incluye 11 ecuaciones diferenciales y 31 constantes biológicas que describen las fases de desarrollo del nematodo y su relación con la bacteria. Para simular el comportamiento real de las poblaciones, las constantes, que representan parámetros biológicos, deberán ser evaluadas en condiciones controladas similares a la del microcosmo del suelo. -

Pale Cyst Nematode Globodera Pallida
Michigan State University’s invasive species factsheets Pale cyst nematode Globodera pallida The pale cyst nematode is a serious pest of potatoes around the world and is a target of strict regulatory actions in the United States. An introduction to Michigan may adversely affect production and marketing of potatoes and other solanaceous crops. Michigan risk maps for exotic plant pests. Other common names potato cyst nematode, white potato cyst nematode, pale potato cyst nematode Systematic position Nematoda > Tylenchida > Heteroderidae > Globodera pallida (Stone) Behrens Global distribution Worldwide distribution in potato-producing regions. Mature females (white) and a cyst (brown) of potato cyst nematode attached to the roots. (Photo: B. Hammeraas, Bioforsk - Norwegian Institute for Agricultural Africa: Algeria, Tunisia; Asia: India, Pakistan, and Environmental Research, Bugwood.org) Turkey; Europe: Austria, Belgium, Bulgaria, Croatia, Czech Republic, Cyprus, Faroe Islands, Finland, pale cyst nematode move to, penetrate and feed on host France, Germany, Greece, Hungary, Iceland, Ireland, roots. After mating, fertilized eggs develop inside females Italy, Luxembourg, Malta, Netherlands, Norway, Poland, that are attached to roots. When females die their skin Portugal, Romania, Spain, Sweden, Switzerland, United hardens and becomes a protective brown cover (cyst) Kingdom; Latin America: Argentina, Bolivia, Chile, around the eggs. Each cyst contains hundreds of eggs, and Colombia, Ecuador, Panama, Peru, Venezuela; Oceania: it can remain viable for many years in the absence of host New Zealand; North America: Idaho, Canada (a small plants. One generation normally occurs during one growing area of Newfoundland). season. Quarantine status Identification In 2006, the pale cyst nematode was detected at At flowering or later stages of a host plant, mature a potato processing facility in Idaho. -

Pasteuria Penetrans COMO AGENTE DE CONTROL BIOLÓGICO DE Meloidogyne Spp
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Publicaciones seriadas del CENSA (E-Journals) Rev. Protección Veg. Vol. 25 No. 3 (2010): 137-149 Artículo Reseña Pasteuria penetrans COMO AGENTE DE CONTROL BIOLÓGICO DE Meloidogyne spp. Lucila Gómez*, Hortensia Gandarilla**, Mayra G. Rodríguez* *Centro Nacional de Sanidad Agropecuaria (CENSA). Apartado 10. San José de las Lajas, La Habana, Cuba. Correo electrónico: [email protected]; **Laboratorio Central de Cuarentena Vegetal (LCCV). Ayuntamiento No 231 e/ Lombillo y San Pedro, Plaza de la Revolución, Cuidad de La Habana, Cuba RESUMEN: El objetivo de esta reseña es hacer una recopilación de la información más reciente de la biología, ecología y potencialidades de Pasteuria penetrans y otras especies del género como agente de control biológico. P. p enet r a ns es una bacteria formadora de endosporas y micelio, parásito obligado de nematodos del género Meloidogyne. En los últimos años muchos han sido los progresos para entender su biología e importancia como agente de control biológico de nematodos formadores de agallas en el suelo. Las especies del género Pasteuria, están ampliamente distribuida a nivel mundial y han sido informadas, en al menos, 80 países infectando 323 especies de nematodos pertenecientes a 116 géneros que incluyen nematodos de vida libre, fitoparásitos y nematodos entomopatógenos. La temperatura y las condiciones físico-químicas, así como los factores bióticos del suelo desempeñan una importante función en su biología y patogenicidad. El cultivo in vitro de la bacteria no ha sido exitoso hasta el presente, por lo que las producciones de endosporas a gran escala se realizan en sistemas in vivo. -

Review on Control Methods Against Plant Parasitic Nematodes Applied in Southern Member States (C Zone) of the European Union
agriculture Review Review on Control Methods against Plant Parasitic Nematodes Applied in Southern Member States (C Zone) of the European Union Nicola Sasanelli 1, Alena Konrat 2, Varvara Migunova 3,*, Ion Toderas 4, Elena Iurcu-Straistaru 4, Stefan Rusu 4, Alexei Bivol 4, Cristina Andoni 4 and Pasqua Veronico 1 1 Institute for Sustainable Plant Protection, CNR, St. G. Amendola 122/D, 70126 Bari, Italy; [email protected] (N.S.); [email protected] (P.V.) 2 Federal State Budget Scientific Institution “Federal Scientific Centre VIEV” (FSC VIEV) of RAS, Bolshaya Cheryomushkinskaya 28, 117218 Moscow, Russia; [email protected] 3 A.N. Severtsov Institute of Ecology and Evolution, Russian Academy of Sciences, Leninsky Prospect 33, 119071 Moscow, Russia 4 Institute of Zoology, MECC, Str. Academiei 1, 2028 Chisinau, Moldova; [email protected] (I.T.); [email protected] (E.I.-S.); [email protected] (S.R.); [email protected] (A.B.); [email protected] (C.A.) * Correspondence: [email protected] Abstract: The European legislative on the use of different control strategies against plant-parasitic nematodes, with particular reference to pesticides, is constantly evolving, sometimes causing confu- Citation: Sasanelli, N.; Konrat, A.; sion in the sector operators. This article highlights the nematode control management allowed in the Migunova, V.; Toderas, I.; C Zone of the European Union, which includes the use of chemical nematicides (both fumigant and Iurcu-Straistaru, E.; Rusu, S.; Bivol, non-fumigant), agronomic control strategies (crop rotations, biofumigation, cover crops, soil amend- A.; Andoni, C.; Veronico, P. Review ments), the physical method of soil solarization, the application of biopesticides (fungi, bacteria and on Control Methods against Plant their derivatives) and plant-derived formulations. -

8736 Federal Register / Vol
8736 Federal Register / Vol. 77, No. 31 / Wednesday, February 15, 2012 / Rules and Regulations Aureobasidium pullulans strains DSM 5. Fleet, G.H. 2003. Yeast interactions and Dated: January 30, 2012. 14940 and DSM 14941. Therefore, an wine flavor. Int. J. Food Microbiol., 86: Steven Bradbury, exemption from the requirement of a 11–22. Director, Office of Pesticide Programs. tolerance is established for residues of 6. Granado, J., B. Thurig, E. Kieffer, L. Petrini, Aureobasidium pullulans strains DSM A. Fliessbach, L. Tamm, F.P. Weibel, Therefore, 40 CFR chapter I is 14940 and DSM 14941 in or on all food G.S. Wyss. 2008. Culturable fungi of amended as follows: stored ‘Golden Delicious’ apple fruits: a commodities when applied as a PART 180—[AMENDED] preharvest fungicide and used in one-season comparison study of organic and integrated production systems in accordance with good agricultural ■ Switzerland. Microb. Ecol., 56: 720–732. 1. The authority citation for part 180 practices. 7. Gao, X.-X., H. Zhou, D.-Y. Xu, C.-H. Yu, continues to read as follows: IX. Statutory and Executive Order Y.-Q. Chen, L.-H. Qu. 2005. High Authority: 21 U.S.C. 321(q), 346a and 371. Reviews diversity of endophytic fungi from the pharmaceutical plant, Heterosmilax ■ 2. Section 180.1312 is added to This final rule establishes a tolerance japonica Kunth revealed by cultivation- subpart D to read as follows: under section 408(d) of FFDCA in independent approach. FEMS Microbiol. § 180.1312 Aureobasidium pullulans response to a petition submitted to the Letters, 249: 255–266. strains DSM 14940 and DSM 14941; Agency. -

Daphnia Magna Changes Withhostage.Clonaldifferences Make Populations, Ourresultshighlightabroad to Resistitsnaturalbacterialpathogen D
© 2014. Published by The Company of Biologists Ltd | The Journal of Experimental Biology (2014) 217, 3929-3934 doi:10.1242/jeb.111260 RESEARCH ARTICLE The development of pathogen resistance in Daphnia magna: implications for disease spread in age-structured populations Jennie S. Garbutt*, Anna J. P. O’Donoghue, Seanna J. McTaggart, Philip J. Wilson and Tom J. Little ABSTRACT Understanding of the development of the vertebrate immune system Immunity in vertebrates is well established to develop with time, but in early life is centered on the development of the acquired immune the ontogeny of defence in invertebrates is markedly less studied. system. It is generally accepted that, even as acquired immunity Yet, age-specific capacity for defence against pathogens, coupled remains undeveloped or under-developed, young individuals will have with age structure in populations, has widespread implications for full use of their innate immune system. With this in mind, it is disease spread. Thus, we sought to determine the susceptibility of tempting to assume that invertebrates, which possess only an innate hosts of different ages in an experimental invertebrate immune system, would not show an immune system or defence host–pathogen system. In a series of experiments, we show that the ontogeny similar to that of young vertebrates. Yet, in general, the ability of Daphnia magna to resist its natural bacterial pathogen innate immune system of invertebrates is surprisingly capable, and Pasteuria ramosa changes with host age. Clonal differences make there is even evidence that it develops through time in response to it difficult to draw general conclusions, but the majority of challenges from pathogens (McTaggart et al., 2012), or can be observations indicate that resistance increases early in the life of D. -

Potato Cyst Nematodes Factsheet
Biosecurity SA – Plant Health Exotic Plant Pest Hotline: 1800 084 881 (available 24 hours) Email [email protected] June 2017 Potato cyst nematodes Globodera pallida and Globodera rostochiensis WHAT IS IT? Pale potato cyst nematode (Globodera pallida) is an exotic plant pest not present in Australia Golden potato cyst nematode (Globodera rostochiensis) is present in some areas of Victoria but is not in South Australia. These nematodes are a serious threat to Australia’s potato industry. Potato cyst nematodes (PCN) are soil-borne microscopic worms which feed on roots of potato plants. Root development and tuber yield is reduced and plant growth is stunted. The biology and symptoms caused by both species Crop damage caused by PCN. are similar. Photo courtesy© Syngenta 2013 The occurrence of PCN is related to the presence of a host and not to soil type or soil temperature. The preferred host of PCN is potato. PCN can infest plants such as tomato, eggplant and some solanaceous weeds. PCN can survive as cysts in the soil for many years in the absence of host plants. PCN is subject to stringent quarantine and regulatory procedures wherever it occurs. WHAT TO LOOK FOR? The symptoms of attack by Globodera species are not specific. Symptoms may appear similar to water or nutrient deficiencies or wilt diseases. Infested potato plants have a reduced root system which is abnormally branched and brownish in colour. Growth is stunted, leaves yellow early or turn a dull colour, flowering is delayed and plants may wilt. At or after flowering very tiny white, yellow or brown cysts about the size of a pin head (0.5 mm) might be seen on the outside of roots HOW DOES IT SURVIVE? PCN are small worms less than 1 mm in size.