And Post-Synaptic Abnormalities in Schizophrenia Lynsey S Hall
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Genetic Variants Contribute to Gene Expression Variability in Humans
INVESTIGATION Genetic Variants Contribute to Gene Expression Variability in Humans Amanda M. Hulse* and James J. Cai*,†,1 *Interdisciplinary Program in Genetics and †Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, Texas 77843-4458 ABSTRACT Expression quantitative trait loci (eQTL) studies have established convincing relationships between genetic variants and gene expression. Most of these studies focused on the mean of gene expression level, but not the variance of gene expression level (i.e., gene expression variability). In the present study, we systematically explore genome-wide association between genetic variants and gene expression variability in humans. We adapt the double generalized linear model (dglm) to simultaneously fit the means and the variances of gene expression among the three possible genotypes of a biallelic SNP. The genomic loci showing significant association between the variances of gene expression and the genotypes are termed expression variability QTL (evQTL). Using a data set of gene expression in lymphoblastoid cell lines (LCLs) derived from 210 HapMap individuals, we identify cis-acting evQTL involving 218 distinct genes, among which 8 genes, ADCY1, CTNNA2, DAAM2, FERMT2, IL6, PLOD2, SNX7, and TNFRSF11B, are cross-validated using an extra expression data set of the same LCLs. We also identify 300 trans-acting evQTL between .13,000 common SNPs and 500 randomly selected representative genes. We employ two distinct scenarios, emphasizing single-SNP and multiple-SNP effects on expression variability, to explain the formation of evQTL. We argue that detecting evQTL may represent a novel method for effectively screening for genetic interactions, especially when the multiple-SNP influence on expression variability is implied. -

Retinoic Acid Induced 1 Gene Analysis in Humans and Zebrafish
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2009 RETINOIC ACID INDUCED 1 GENE ANALYSIS IN HUMANS AND ZEBRAFISH Bijal Vyas Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medical Genetics Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/1901 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Bijal Akshay Vyas, 2009 All Rights Reserved RETINOIC ACID INDUCED 1 GENE ANALYSIS IN HUMANS AND ZEBRAFISH A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by BIJAL AKSHAY VYAS Bachelor of Biology North Carolina State University, 2007 Director: SARAH H. ELSEA Associate Professor, Department of Human and Molecular Genetics Department of Pediatrics Virginia Commonwealth University Richmond, Virginia August, 2009 Acknowledgement I would like to thank all the people who have supported and motivated me throughout my graduate studies. I had the great opportunity to work in Dr. Sarah Elsea‘s lab and I sincerely thank her for her willingness to help, patience and encouraging me after my failed experiments. I am also greatly thankful to Dr. Jim Lister for guiding and helping me every step of the way in finishing my zebrafish project. I also thank my third committee member, Dr. Andrew Davies for his project consultations, helpful alternatives and encouragement. -

Protein Interaction Network of Alternatively Spliced Isoforms from Brain Links Genetic Risk Factors for Autism
ARTICLE Received 24 Aug 2013 | Accepted 14 Mar 2014 | Published 11 Apr 2014 DOI: 10.1038/ncomms4650 OPEN Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism Roser Corominas1,*, Xinping Yang2,3,*, Guan Ning Lin1,*, Shuli Kang1,*, Yun Shen2,3, Lila Ghamsari2,3,w, Martin Broly2,3, Maria Rodriguez2,3, Stanley Tam2,3, Shelly A. Trigg2,3,w, Changyu Fan2,3, Song Yi2,3, Murat Tasan4, Irma Lemmens5, Xingyan Kuang6, Nan Zhao6, Dheeraj Malhotra7, Jacob J. Michaelson7,w, Vladimir Vacic8, Michael A. Calderwood2,3, Frederick P. Roth2,3,4, Jan Tavernier5, Steve Horvath9, Kourosh Salehi-Ashtiani2,3,w, Dmitry Korkin6, Jonathan Sebat7, David E. Hill2,3, Tong Hao2,3, Marc Vidal2,3 & Lilia M. Iakoucheva1 Increased risk for autism spectrum disorders (ASD) is attributed to hundreds of genetic loci. The convergence of ASD variants have been investigated using various approaches, including protein interactions extracted from the published literature. However, these datasets are frequently incomplete, carry biases and are limited to interactions of a single splicing isoform, which may not be expressed in the disease-relevant tissue. Here we introduce a new interactome mapping approach by experimentally identifying interactions between brain-expressed alternatively spliced variants of ASD risk factors. The Autism Spliceform Interaction Network reveals that almost half of the detected interactions and about 30% of the newly identified interacting partners represent contribution from splicing variants, emphasizing the importance of isoform networks. Isoform interactions greatly contribute to establishing direct physical connections between proteins from the de novo autism CNVs. Our findings demonstrate the critical role of spliceform networks for translating genetic knowledge into a better understanding of human diseases. -

Par6c Is at the Mother Centriole and Controls Centrosomal Protein
860 Research Article Par6c is at the mother centriole and controls centrosomal protein composition through a Par6a-dependent pathway Vale´rian Dormoy, Kati Tormanen and Christine Su¨ tterlin* Department of Developmental and Cell Biology, University of California, Irvine, Irvine, CA 92697-2300, USA *Author for correspondence ([email protected]) Accepted 3 December 2012 Journal of Cell Science 126, 860–870 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.121186 Summary The centrosome contains two centrioles that differ in age, protein composition and function. This non-membrane bound organelle is known to regulate microtubule organization in dividing cells and ciliogenesis in quiescent cells. These specific roles depend on protein appendages at the older, or mother, centriole. In this study, we identified the polarity protein partitioning defective 6 homolog gamma (Par6c) as a novel component of the mother centriole. This specific localization required the Par6c C-terminus, but was independent of intact microtubules, the dynein/dynactin complex and the components of the PAR polarity complex. Par6c depletion resulted in altered centrosomal protein composition, with the loss of a large number of proteins, including Par6a and p150Glued, from the centrosome. As a consequence, there were defects in ciliogenesis, microtubule organization and centrosome reorientation during migration. Par6c interacted with Par3 and aPKC, but these proteins were not required for the regulation of centrosomal protein composition. Par6c also associated with Par6a, which controls protein recruitment to the centrosome through p150Glued. Our study is the first to identify Par6c as a component of the mother centriole and to report a role of a mother centriole protein in the regulation of centrosomal protein composition. -

Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17P11.2 and the Syntenic Region of the Mouse
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Article Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17p11.2 and the Syntenic Region of the Mouse Weimin Bi,1,6 Jiong Yan,1,6 Paweł Stankiewicz,1 Sung-Sup Park,1,7 Katherina Walz,1 Cornelius F. Boerkoel,1 Lorraine Potocki,1,3 Lisa G. Shaffer,1 Koen Devriendt,4 Małgorzata J.M. Nowaczyk,5 Ken Inoue,1 and James R. Lupski1,2,3,8 Departments of 1Molecular & Human Genetics, 2Pediatrics, Baylor College of Medicine, 3Texas Children’s Hospital, Houston, Texas 77030, USA; 4Centre for Human Genetics, University Hospital Gasthuisberg, Catholic University of Leuven, B-3000 Leuven, Belgium; 5Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario L8S 4J9, Canada Smith-Magenis syndrome (SMS) is a multiple congenital anomaly/mental retardation syndrome associated with behavioral abnormalities and sleep disturbance. Most patients have the same ∼4 Mb interstitial genomic deletion within chromosome 17p11.2. To investigate the molecular bases of the SMS phenotype, we constructed BAC/PAC contigs covering the SMS common deletion interval and its syntenic region on mouse chromosome 11. Comparative genome analysis reveals the absence of all three ∼200-kb SMS-REP low-copy repeats in the mouse and indicates that the evolution of SMS-REPs was accompanied by transposition of adjacent genes. Physical and genetic map comparisons in humans reveal reduced recombination in both sexes. Moreover, by examining the deleted regions in SMS patients with unusual-sized deletions, we refined the minimal Smith-Magenis critical region (SMCR) to an ∼1.1-Mb genomic interval that is syntenic to an ∼1.0-Mb region in the mouse. -

Targeting Hedgehog Signalling Through the Ubiquitylation Process: the Multiple Roles of the HECT-E3 Ligase Itch
cells Review Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch Paola Infante 1,†, Ludovica Lospinoso Severini 2,† , Flavia Bernardi 2, Francesca Bufalieri 2 and Lucia Di Marcotullio 2,3,* 1 Center for Life NanoScience@Sapienza, Istituto Italiano di Tecnologia, 00161 Rome, Italy; [email protected] 2 Department of Molecular Medicine, University of Rome La Sapienza, 00161 Rome, Italy; [email protected] (L.L.S.); fl[email protected] (F.B.); [email protected] (F.B.) 3 Istituto Pasteur-Fondazione Cenci Bolognetti, University of Rome La Sapienza, 00161 Rome, Italy * Correspondence: [email protected]; Tel.: +39-06-49255657 † These authors contributed equally to this work. Received: 28 December 2018; Accepted: 26 January 2019; Published: 29 January 2019 Abstract: Hedgehog signalling (Hh) is a developmental conserved pathway strongly involved in cancers when deregulated. This important pathway is orchestrated by numerous regulators, transduces through distinct routes and is finely tuned at multiple levels. In this regard, ubiquitylation processes stand as essential for controlling Hh pathway output. Although this post-translational modification governs proteins turnover, it is also implicated in non-proteolytic events, thereby regulating the most important cellular functions. The HECT E3 ligase Itch, well known to control immune response, is emerging to have a pivotal role in tumorigenesis. By illustrating Itch specificities on Hh signalling key components, here we review the role of this HECT E3 ubiquitin ligase in suppressing Hh-dependent tumours and explore its potential as promising target for innovative therapeutic approaches. Keywords: Hedgehog; cancer; ubiquitylation; Itch; Numb; β-arrestin2; GLI1; SuFu; Patched1 1. -

FMRP Links Optimal Codons to Mrna Stability in Neurons
FMRP links optimal codons to mRNA stability in neurons Huan Shua,1, Elisa Donnardb, Botao Liua, Suna Junga, Ruijia Wanga, and Joel D. Richtera aProgram in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605; and bBioinformatics and Integrative Biology, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Lynne E. Maquat, University of Rochester School of Medicine and Dentistry, Rochester, NY, and approved October 12, 2020 (received for review May 8, 2020) Fragile X syndrome (FXS) is caused by inactivation of the FMR1 Here, we have used ribosome profiling and RNA sequencing gene and loss of encoded FMRP, an RNA binding protein that re- (RNA-seq) to investigate translational dysregulation in the presses translation of some of its target transcripts. Here we use FMRP KO cortex and found that FMRP coordinates the link ribosome profiling and RNA sequencing to investigate the dysre- between RNA destruction and codon usage bias (codon opti- gulation of translation in the mouse brain cortex. We find that mality). We find that the apparent dysregulation of translational most changes in ribosome occupancy on hundreds of mRNAs are activity (i.e., ribosome occupancy) in FMRP KO cortex can be largely driven by dysregulation in transcript abundance. Many accounted for by commensurate changes in steady-state RNA down-regulated mRNAs, which are mostly responsible for neuro- levels. Down-regulated mRNAs in FMRP KO cortex are nal and synaptic functions, are highly enriched for FMRP binding enriched for those that encode factors involved in neuronal and targets. RNA metabolic labeling demonstrates that, in FMRP- synaptic functions and are highly enriched for FMRP binding deficient cortical neurons, mRNA down-regulation is caused by targets. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Comparison of Orthologs Across Multiple Species by Various Strategies
COMPARISON OF ORTHOLOGS ACROSS MULTIPLE SPECIES BY VARIOUS STRATEGIES BY HUI LIU DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biophysics and Computational Biology in the Graduate College of the University of Illinois at Urbana-Champaign, 2014 Urbana, Illinois Doctoral Committee: Professor Eric Jakobsson, Chair, Director of Research Professor Gene E. Robinson Associate Professor Saurabh Sinha Assistant Professor Jian Ma Abstract Thanks to the improvement of genome sequencing technology, abundant multi-species genomic data now became available and comparative genomics continues to be a fast prospering filed of biological research. Through the comparison of genomes of different organisms, we can understand what, at the molecular level, distinguishes different life forms from each other. It shed light on revealing the evolution of biology. And it also helps to refine the annotations and functions of individual genomes. For example, through comparisons across mammalian genomes, we can give an estimate of the conserved set of genes across mammals and correspondingly, find the species-specific sets of genes or functions. However, comparative genomics can be feasible only if a meaningful classification of genes exists. A natural way to do so is to delineate sets of orthologous genes. However, debates exist about the appropriate way to define orthologs. It is originally defined as genes in different species which derive from speciation events. But such definition is not sufficient to derive orthologous genes due to the complexity of evolutionary events such as gene duplication and gene loss. While it is possible to correctly figure out all the evolutionary events with the true phylogenetic tree, the true phylogenetic tree itself is impractical to be inferred. -

TRIM37 Orchestrates Renal Cell Carcinoma Progression Via Histone
Miao et al. Journal of Experimental & Clinical Cancer Research (2021) 40:195 https://doi.org/10.1186/s13046-021-01980-0 RESEARCH Open Access TRIM37 orchestrates renal cell carcinoma progression via histone H2A ubiquitination- dependent manner Chenkui Miao1†, Chao Liang1†,PuLi1†, Bianjiang Liu1, Chao Qin1, Han Yuan2, Yiyang Liu1, Jundong Zhu3, Yankang Cui1, Aiming Xu1, Shangqian Wang1, Shifeng Su1, Jie Li1, Pengfei Shao1* and Zengjun Wang1* Abstract Background: Ubiquitylation modification is one of the multiple post-transcriptional process to regulate cellular physiology, including cell signaling, cycle regulation, DNA repair and transcriptional regulation. Members of TRIM family proteins could be defined as E3 ubiquitin ligases as they contain a RING-finger domain, and alterations of TRIM proteins are involved into a broad range of diverse disorders including cancer. TRIM37 is a novel discovered E3 ubiquitin ligase and acts as a oncoprotein in multiple human neoplasms, however its biological role in RCC still remains elusive. Methods: RCC microarray chips and public datasets were screened to identify novel TRIMs member as TRIM37, which was dysregulated in RCC. Gain or loss of functional cancer cell models were constructed, and in vitro and in vivo assays were performed to elucidate its tumorigenic phenotypes. Interactive network analyses were utilized to define intrinsic mechanism. Results: We identified TRIM37 was upregulated in RCC tumors, and its aberrant function predicted aggressive neoplastic phenotypes, poorer survival endings. TRIM37 promoted RCC cells EMT and malignant progression via TGF-β1 signaling activation, as a consequence of directly mediated by ubiquitinating-H2A modifications. Conclusions: Our findings identified a previously unappreciated role of TRIM37 in RCC progression and prognostic prediction. -
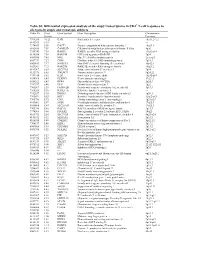
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3