Titelblad Engels 2006
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Zirconium Phosphate/Polycation Thin Films Grown by Sequential Adsorption Reactions
1414 Chem. Mater. 1997, 9, 1414-1421 Characterization of Zirconium Phosphate/Polycation Thin Films Grown by Sequential Adsorption Reactions Hyuk-Nyun Kim, Steven W. Keller,† and Thomas E. Mallouk* Department of Chemistry, Pennsylvania State University, University Park, Pennsylvania 16802 Johannes Schmitt Institut fu¨ r Physikalische Chemie, Johannes Gutenberg-Universita¨t, D-55099 Mainz, Germany Gero Decher Institut Charles Sadron, Universite´ Louis Pasteur, F-67083 Strasbourg Cedex, France Received January 6, 1997. Revised Manuscript Received April 7, 1997X Monolayer and multilayer thin films consisting of anionic R-zirconium phosphate (R-ZrP) sheets and polycations (poly(allylamine hydrochloride) (PAH), cytochrome c) were character- ized by transmission electron microscopy (TEM), ellipsometry, UV-visible absorbance spectroscopy, reflectance FT-IR, XPS, and X-ray diffraction. Titration and powder X-ray diffraction experiments confirm that exfoliation of R-ZrP begins to occur when enough tetra- (n-butylammonium) hydroxide (TBA+OH-) has been added to exceed single-layer packing + + - of TBA ions (x 0.50) in the intercalation compound Zr(HPO4)2-x(TBA PO4 )x‚nH2O. The identical contrast≈ of many sheets in TEM micrographs suggests that the suspension is unilamellar. Alternately dipping cationic substrates into R-ZrP-containing suspensions and aqueous PAH gives a multilayer film that resembles the corresponding bulk intercalation compound. X-ray photoelectron spectra of multilayer films show that they are Zr-rich, relative to R-ZrP, consistent with some corrosion during the exfoliation reaction. The R-ZrP/ PAH layer pair thickness is 13/14.7 Å, as measured by ellipsometry/X-ray diffraction, respectively. A 13-layer pair film is sufficiently well-ordered in the stacking direction to give a Bragg peak in the diffraction pattern. -

Monolithic Cells for Solar Fuels Chem Soc Rev
Volume 43 Number 23 7 December 2014 Pages 7957–8194 Chem Soc Rev Chemical Society Reviews www.rsc.org/chemsocrev Includes a collection of articles on the theme of CO2 Capture and Chemical Recycling ISSN 0306-0012 TUTORIAL REVIEW Johan A. Martens et al. Monolithic cells for solar fuels Chem Soc Rev View Article Online TUTORIAL REVIEW View Journal | View Issue Monolithic cells for solar fuels† a a b c d Cite this: Chem. Soc. Rev., 2014, Jan Ronge´, Tom Bosserez, David Martel, Carlo Nervi, Luca Boarino, a b c a 43,7963 Francis Taulelle, Gero Decher, Silvia Bordiga and Johan A. Martens* Hybrid energy generation models based on a variety of alternative energy supply technologies are considered the best way to cope with the depletion of fossil energy resources and to limit global warming. One of the currently missing technologies is the mimic of natural photosynthesis to convert carbon dioxide and water into chemical fuel using sunlight. This idea has been around for decades, but artificial photosynthesis of organic molecules is still far away from providing real-world solutions. The scientific challenge is to perform in an efficient way the multi-electron transfer reactions of water oxidation and carbon dioxide reduction using holes and single electrons generated in an illuminated semiconductor. In this tutorial review the design of photoelectrochemical (PEC) cells that combine solar water oxidation and CO2 reduction is discussed. In such PEC cells simultaneous transport and efficient use of light, electrons, protons and molecules has to be managed. It is explained how efficiency can be Creative Commons Attribution-NonCommercial 3.0 Unported Licence. -

AND MICROCARRIER FABRICATION a Thes
A NOVEL μ-FLUIDIC CHANNEL ASSISTED ENCAPSULATION TECHNIQUE FOR LAYER-BY-LAYER POLYMER NANO- AND MICROCARRIER FABRICATION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Jingyu Li August, 2015 A NOVEL μ-FLUIDIC CHANNEL ASSISTED ENCAPSULATION TECHNIQUE FOR LAYER-BY-LAYER POLYMER NANO- AND MICROCARRIER FABRICATION Jingyu Li Thesis Approved: Accepted: Advisor Department Chair Dr. Younjin Min Dr. Sadhan C. Jana Committee Member Dean of the College Dr. Mukerrem Cakmak Dr. Eric J. Amis Committee Member Interim Dean of the Graduate School Dr. Hossein Tavana Dr. Rex D. Ramsier Date ii ABSTRACT Layer-by-layer (LbL) assembly is a popular technique for fabricating thin multilayer films on templates by depositing alternating layers of oppositely charged polyelectrolytes with rinsing steps in between. The LbL technique fabricated capsules can be used for biomedical applications such as drug and vaccine delivery, biosensors and bioreactors. LbL assembly enables us to use various types, sizes, and shapes of particles as templates, like silica particles, calcium carbonate particles and metal particles. In addition, a suite of water-soluble polymers with different properties can be applied to LbL assembly to meet researchers` requirements. Due to accurate control on size and shape, LbL technique is capable of fabricating small particles below 200 nm, which is in the optimal size for drug carriers and small enough to avoid clogging blood capillaries. The versatility of nano- and microcapsules has captured the attention of researchers to develop fabrication methods of LbL particles. The traditional LbL assembly method has simple process, but which is inefficient and has limitations due to repeated centrifugation steps. -

1 Layer-By-Layer Assembly (Putting Molecules to Work) Gero Decher
j1 1 Layer-by-Layer Assembly (Putting Molecules to Work) Gero Decher 1.1 The Whole is More than the Sum of its Parts The properties of a material arise from the structural arrangement of its constituents and their dynamics. In nature, a hierarchical organization assures that a required property of a material is realized with a minimum of constituents and a minimum of complexity through processes of evolutionary selection. In the living world functional entities exist on any length scale ranging from atoms to whole organisms, complex properties of biological materials such as cellular life emerging at the upper end of the nanoscale (Figure 1.1). Chemistry, Physics and Materials Science are increasingly approaching maturity on the molecular and macroscopic length scales, shifting importance toward new nanoscale materials and nanoorganized systems. New materials properties, a pre- requisite for responding to the emerging problems of the world population, will require materials design at the nanoscale, forcing the development of new multi- material nanocomposites. In complex systems, new properties appear that are not observed for each individual component. While it is trivial that electrons and nuclei form atoms (sub-Angstrom scale), that atoms form molecules (Angstrom scale) or that monomers can be transformed into polymers (early nanometer scale), we are just beginning to explore the potential of supramolecular assemblies of multifunctional objects. Figure 1.1 summarizes how complex materials properties evolve hierarchically over a length scale of several orders of magnitude. 1.2 From Self-Assembly to Directed Assembly Imagine a self-assembly experiment involving several different chemical species and obtaining equilibrium. -
Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials Gero Decher (Editor), Joe B
To purchase this product, please visit https://www.wiley.com/en-us/9783527605415 Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials Gero Decher (Editor), Joe B. Schlenoff (Editor) E-Book 978-3-527-60541-5 March 2006 $205.00 DESCRIPTION Materials scientists are often faced with the problem of modifying surfaces of objects, yet keeping their shape and properties. This book provides a detailed survey on the new technology of adsorption from solution for the fabrication of molecularly ordered multicomposite films in order to replace and expand on the well known Langmuir-Blodgett technology and to open the field of molecular self-assembly to materials and biosciences. The book is aimed at scientists who want to integrate several different functional entities in a single device. To this audience it presents the technique of layer-by-layer assembly as today's most powerful key technology, which is low cost, solution based and very robust. It is already beginning to make the transition from academic research into industrial mass production. ABOUT THE AUTHOR Gero Decher is a Distinguished Professor of Chemistry at the University of Strasbourg, France, a senior member of the Institut Universitaire de France and a member of the International Center for Frontier Research in Chemistry. His research team is located at CNRS Institut Charles Sadron in Strasbourg where he continues to develop the layer-by-layer assembly method in collaboration with his colleagues, Pierre Schaaf and Jean-Claude Voegel. This method is applied in many laboratories worldwide in various scientific disciplines, including chemistry, materials science and biotechnology. Gero Decher has received numerous awards, including the ECIS-Rhodia prize in 2010 and the Grand Prix of the French "Academie des Sciences" for Nanobiotechnology in 2009. -

Department of Biotechnology Course Contents & Syllabus
18th BoS, Biotechnology (Item 18.19) dt. 23.05.2020 Department of Biotechnology Course Contents & Syllabus Hemvati Nandan Bahuguna Garhwal University (A Central University) Srinagar, Garhwal, 246 174, Uttarakhand M.Sc. Biotechnology, Course Contents & Syllabus- effective from July 2020 (1) 18th BoS, Biotechnology (Item 18.19) dt. 23.05.2020 M. Sc. Biotechnology ( E ffective from J u l y 2 0 2 0 ) Code Course Contents L T P C M.M Semester I (July to November) S0LS/MBT/C0001 Biochemistry 3 0 0 3 100 S0LS/MBT/C0002 Cell Biology & Membrane Biophysics 3 0 0 3 100 S0LS/MBT/C0003 Molecular Biology & Genetics 3 0 0 3 100 S0LS/MBT/C0004 Bio-Analytical Techniques 3 0 0 3 100 S0LS/MBT/C0005 Lab Course based on course C0001 & C0002 0 0 3 3 100 S0LS/MBT/C0006 Lab Course based on course C0003 & C0004 0 0 3 3 100 Core Credits= 18 600 Semester II (December to April) S0LS/MBT/C0007 Immunology 3 0 0 3 100 S0LS/MBT/C0008 Microbiology & Microbial Genetics 3 0 0 3 100 S0LS/MBT/C0009 Genetic Engineering & Applications 3 0 0 3 100 S0LS/MBT/C0010 Biostatistics & Bioinformatics 3 0 0 3 100 S0LS/MBT/C0011 Lab Course based on course C0007 & C0008 0 0 3 3 100 S0LS/MBT/C0012 Lab Course based on course C0009 & C0010 0 0 3 3 100 S0LS/MBT/SS001 Epigenetics & Cancer Biology 0 0 0 3 100 S0LS/MBT/SS002 Biomedical Technology Core Credits = 18 600 Semester III (July to November) S0LS/MBT/C0013 Plant Biotechnology 3 0 0 3 100 S0LS/MBT/C0014 Intellectual Property Rights, Bioethics, Bio-Entrepreneurship 3 0 0 3 100 S0LS/MBT/C0015 Lab Course based on course C0013 & C0014 0 0 -

Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites Gero Decher, Et Al
Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites Gero Decher, et al. Science 277, 1232 (1997); DOI: 10.1126/science.277.5330.1232 This copy is for your personal, non-commercial use only. If you wish to distribute this article to others, you can order high-quality copies for your colleagues, clients, or customers by clicking here. Permission to republish or repurpose articles or portions of articles can be obtained by following the guidelines here. The following resources related to this article are available online at www.sciencemag.org (this information is current as of February 24, 2010 ): Updated information and services, including high-resolution figures, can be found in the online version of this article at: http://www.sciencemag.org/cgi/content/full/277/5330/1232 This article has been cited by 30 articles hosted by HighWire Press; see: http://www.sciencemag.org/cgi/content/full/277/5330/1232#otherarticles This article appears in the following subject collections: Materials Science http://www.sciencemag.org/cgi/collection/mat_sci on February 24, 2010 www.sciencemag.org Downloaded from Science (print ISSN 0036-8075; online ISSN 1095-9203) is published weekly, except the last week in December, by the American Association for the Advancement of Science, 1200 New York Avenue NW, Washington, DC 20005. Copyright 1997 by the American Association for the Advancement of Science; all rights reserved. The title Science is a registered trademark of AAAS. Physics (Cornell Univ. Press, Ithaca, NY, 1979); F. W. 27. J. T. Chen, E. L. Thomas, C. K. Ober, G.-P. Mao, 32. We have benefited from interactions with many stu- Wiegel, Conformational Phase Transitions in a Mac- ibid. -

Unesco – Eolss Sample Chapters
MEDICAL SCIENCES - Vol. II – Nanobiotechnology - C. Ruggiero NANOBIOTECHNOLOGY C. Ruggiero Department of Communication Computer and System Sciences, University of Genoa, Italy Keywords: Microtechnology, Nanotechnology, Nanoscale structures, Tunnelling Microscope, Biology, Medicine, Nanobiotechnology, biomolecules, nanoparticles, ultra thin films,Carbon Nanotubes, Genomic, Proteomic Analysis, DNA Microarrays,Protein Antibody Microarrays, Nanoparticles , Langmuir Blodgett Techniques , 2 Biomedical Applications, Sensors, Radiation oncology Contents 1. Microtechnology and Nanotechnology 2. The dawning of Nanotechnology 3. Nanoscale Structures: Technology and Applications 4. A key instrument for Nanotechnology: the Scanning Tunnelling Microscope 5. Nanotechnology and government policies 6. The Impact of Nanotechnology on Biology and Medicine: Nanobiotechnology 7. Assemblies of Organized Biomolecules and Nanoparticles: Ultra Thin Films 7.1 Langmuir Blodgett Techniques 7.2 Processing from A Solution: Spin Coating and Solution Casting 7.3 Chemical Self-Assembly 7.4 Layer-By-Layer Self-Assembly 8. Carbon Nanotubes 8.1 Properties of CNTs 8.2 Biomedical Applications of Cnts 8.2.1 Sensors 8.2.2 Drug Delivery 8.2.3 Implantable Nanorobot, Nanosensors and Devices 8.2.4 Radiation oncology 9. High-Throughput Genomic and Proteomic Analysis: DNA Microarrays and Protein Antibody Microarrays 9.1 DNA Microarrays 9.2 Protein and Antibody Microarrays 10. NanoparticlesUNESCO – EOLSS 10.1 Preparation of Nanoparticles 10.2. The ArtificialSAMPLE Cell CHAPTERS Bibliography 1. Microtechnology and Nanotechnology Since the invention of the transistor in 1947, a miniaturization trend in electronic systems started and will surely continue. This miniaturization of electronic systems, aiming to include increasingly complex functions in limited space and with minimum weight, has originated an increase in information density- a key point in our “Information Society” in which information and communication technologies are ©Encyclopedia of Life Support Systems (EOLSS) MEDICAL SCIENCES - Vol. -

Triblock Copolymers
BIOMIMETIC SUPERSTRUCTURES FROM AMPHIPHILIC ABA - TRIBLOCK COPOLYMERS Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophich-Naturwissenschaftlichen Fakultät der Universität Basel von Julie GRUMELARD aus Saint-Louis, Frankreich BASEL, 2004 Genehmigt von der Philosophich-Naturwissenschaftlichen Fakultät Auf Antrag der Herren Prof. Dr. Wolfgang Meier (Universität Basel) Prof. Dr. Marcus Textor (ETH Zurich) Prof. Dr. Hanspeter Huber (Universität Basel) Basel, den 21.Oktober 2004 Prof. Dr. Hans-Jakob Wirz Dekan 2 « La science restera toujours la satisfaction du plus haut désir de notre nature, la curiosité; elle fournira à l'homme le seul moyen qu'il ait pour améliorer son sort. Elle préserve de l'erreur plutôt qu'elle ne donne la vérité; mais c'est déjà quelque chose d'être sûr de n'être pas dupe. » Ernest RENAN, L'Avenir de la science, Pensées de 1848 (1890), Flammarion 1995 3 Table of contents 1 ABSTRACT 6 2 ABBREVIATIONS 8 3 INTRODUCTION 9 BIOLOGICAL MEMBRANES 9 SYNTHETIC AMPHIPHILES AS BIOMEMBRANE MIMICS 10 PMOXA-PDMS-PMOXA ABA-TRIBLOCK COPOLYMERS 11 AIM OF THE PHD THESIS 14 4 NANOVESICLES 16 4.1 ETHANOL METHOD 16 4.2 DETERGENT METHOD 32 4.2.1 DIALYSIS 34 4.2.2 DILUTION METHOD 36 4.2.3 BIO-BEADS METHOD 38 4.2.4 MEMBRANE PROTEIN RECONSTITUTION 45 4.3 BULK SWELLING 50 4.4 REMINDER OF MAIN RESULTS 53 5 NANOTUBES 54 5.1 BULK DISSOLUTION OR BULK SWELLING 54 5.2 FILM REHYDRATION OR FILM SWELLING 57 5.3 CRYO-TEM 65 5.4 FLUORIMETRY 70 5.5 GOLD LOADED NANOTUBES 72 5.5.1 GOLD PARTICLES ENCAPSULATION -

Multilayer Thin Films
Multilayer Thin Films Sequential Assembly of Nanocomposite Materials Edited by Gero Decher, Joseph B. Schlenoff Multilayer Thin Films Sequential Assembly of Nanocomposite Materials Edited by G. Decher, J.B. Schlenoff Multilayer Thin Films Sequential Assembly of Nanocomposite Materials Edited by Gero Decher, Joseph B. Schlenoff Gero Decher n This book was carefully produced. Nevertheless, Institut Charles Sadron editors, authors and publisher do not warrant the 6, rue Boussingault information contained therein to be free of er- F-67083 Strasbourg Cedex rors. Readers are advised to keep in mind that France statements, data, illustrations, procedural details or other items may inadvertently be inaccurate. Joseph B. Schlenoff Florida State University Library of Congress Card No.: applied for Dept. of Chemistry and Biochemistry Tallahassee, Florida 32306-4390 British Library Cataloguing-in-Publication Data USA A catalogue record for this book is available from the British Library. Bibliographic information published by Die Deutsche Bibliothek Die Deutsche Bibliothek lists this publication in the Deutsche Nationalbibliografie; detailed biblio- graphic data is available in the Internet at http://dnb.ddb.de © 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim All rights reserved (including those of translation in other languages). No part of this book may be reproduced in any form – by photoprinting, mi- crofilm, or any other means – nor transmitted or translated into machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law. Printed in the Federal Republic of Germany Printed on acid-free paper Typesetting K+V Fotosatz GmbH, Beerfelden Printing Strauss Offsetdruck GmbH, Mörlenbach Bookbinding J. -

A Way of Targeting and Detection of Cancer Cells
Seminar – 4. letnik NANOMEDICINE: A WAY OF TARGETING AND DETECTION OF CANCER CELLS Author: Maja Požar Mentor: prof. dr. Rudolf Podgornik Ljubljana, November 2010 ABSTRACT Nanotechnology promises changes in many significant areas of medicine, material science, construction, etc. In medicine advanced nanostructures, such as functional nanoparticles, dendrimers, fullerenes, carbon nanotubes and semiconductor nanocrystals have been exploited for drug delivery, diagnostics and treatment of diseases at the molecular level. Nanoparticles have been developed to improve contrast and allow imaging of target tissues using conventional diagnostic equipment. In this paper we focused mostly on how nanotechnology can change the aspect of targeting and detection of cancer cells and consequently its treatment. Rapid advances in the study of the interface of nanomaterials with biomolecules have led to the programmed self-assembly of nanoparticles through the use of biomolecules. This will result in preserving and improving human health, using molecular knowledge of the human body on nanoscale. INDEX INTRODUCTION ......................................................................................................................................... 2 WHAT IS NANOMEDICINE? ....................................................................................................................... 3 NATURE OF THE NANOWORLD ................................................................................................................. 4 NANOPARTICLES IN CANCER -
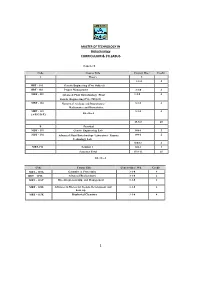
Curriculum & Syllabus 1
MASTER OF TECHNOLOGY IN Biotechnology CURRICULUM & SYLLABUS Semester I Code Course Title Contact Hrs./ Credit A Theory Wk L 3-1-0- 4 MBT - 101 Genetic Engineering (Core Subject) MBT - 102 Project Management 3-1-0 4 MBT - 103 Advanced Plant Biotechnology /Plant 3-1-0 4 Genetic Engineeri ng (Core Subject) MBT - 104 Numerical Analysis and Biostatistics / 3-1-0 4 Mathematics and Biostatistic s MBT - 115 3-1-0 4 Elective-I (A/B/C/D/E) 15-5-0 20 B Practic al MBT - 191 Genetic Engineering Lab 0-0-6 2 MBT - 192 Advanced Plant Biotechnology Laboratory / Enzyme 0-0-6 2 Technology Lab 0-0-12 4 MBT-193 Seminar I 0-0-3 1 Semester Total 15-8-12 25 Elective-I Code Course Title Contact Hrs./ Wk Cre dit MBT – 115A Genomics & Proteomics 3-1-0 4 MBT – 115B Advanced Biochemistry 3-1-0 4 MBT – 115C Bio-entrepreneurship and Management 3-1-0 4 MBT - 115D Advances in Bioreactor Design, Development and 3-1-0 4 Scale up MBT – 115E Biophysical Chemistry 3-1-0 4 1 MASTER OF TECHNOLOGY IN Biotechnology CURRICULUM & SYLLABUS Semester: II Code Course Title Contact Hrs./ Wk Cre dit A Theory L-T-P MBT -201 Advanced Bioinformatics (Core Subjec t) 3-1-0 4 MBT -202 Immunology (Core Subject) 3-1-0 4 MBT -203(A/B) Bioprocess Engineering & Technology / 3-1-0 4 Bio-processing Tec hnology MBT -204 Downstream Processing 3-1-0 4 MBT - 215 3-1-0 4 (A/B/C/D/E/F/G) Elective-II 15-5-0 20 B Practic al MBT - 291 Bioinformatics Laboratory 0-0-6 2 MBT – Downstream processing Lab/ Immunology 0-0-6 2 292(A/B/C) Lab / Animal Cell Culture Laboratory 0-0-12 4 MBT-294 Seminar II 0-0-3 1 Semester