Study Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
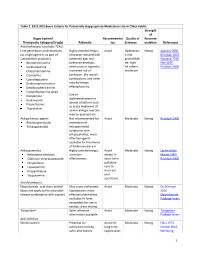
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Drugs to Avoid in Patients with Dementia
Detail-Document #240510 -This Detail-Document accompanies the related article published in- PHARMACIST’S LETTER / PRESCRIBER’S LETTER May 2008 ~ Volume 24 ~ Number 240510 Drugs To Avoid in Patients with Dementia Elderly people with dementia often tolerate drugs less favorably than healthy older adults. Reasons include increased sensitivity to certain side effects, difficulty with adhering to drug regimens, and decreased ability to recognize and report adverse events. Elderly adults with dementia are also more prone than healthy older persons to develop drug-induced cognitive impairment.1 Medications with strong anticholinergic (AC) side effects, such as sedating antihistamines, are well- known for causing acute cognitive impairment in people with dementia.1-3 Anticholinergic-like effects, such as urinary retention and dry mouth, have also been identified in drugs not typically associated with major AC side effects (e.g., narcotics, benzodiazepines).3 These drugs are also important causes of acute confusional states. Factors that may determine whether a patient will develop cognitive impairment when exposed to ACs include: 1) total AC load (determined by number of AC drugs and dose of agents utilized), 2) baseline cognitive function, and 3) individual patient pharmacodynamic and pharmacokinetic features (e.g., renal/hepatic function).1 Evidence suggests that impairment of cholinergic transmission plays a key role in the development of Alzheimer’s dementia. Thus, the development of the cholinesterase inhibitors (CIs). When used appropriately, the CIs (donepezil [Aricept], rivastigmine [Exelon], and galantamine [Razadyne, Reminyl in Canada]) may slow the decline of cognitive and functional impairment in people with dementia. In order to achieve maximum therapeutic effect, they ideally should not be used in combination with ACs, agents known to have an opposing mechanism of action.1,2 Roe et al studied AC use in 836 elderly patients.1 Use of ACs was found to be greater in patients with probable dementia than healthy older adults (33% vs. -

Nomination Background: Ketamine Hydrochloride (CASRN: 1867-66-9)
KETAMINE Nomination TABLE OF CONTENTS Page 1.0 BASIS OF NOMINATION 1 2.0 BACKGROUND INFORMATION 1 3.0 CHEMICAL PROPERTIES 2 3.1 Chemical Identification 2 3.2 Physico-Chemical Properties 3 3.3 Purity and Commercial Availability 4 4.0 PRODUCTION PROCESSES AND ANALYSIS 6 5.0 PRODUCTION AND IMPORT VOLUMES 7 6.0 USES 7 7.0 ENVIRONMENTAL OCCURRENCE 7 8.0 HUMAN EXPOSURE 7 9.0 REGULATORY STATUS 7 10.0 CLINICAL PHARMACOLOGY 7 11.0 TOXICOLOGICAL DATA 13 11.1 General Toxicology 13 11.2 Neurotoxicology 14 12.0 CONCLUSIONS 15 APPENDIX A 17 APPENDIX B 23 APPENDIX C 27 1 KETAMINE 1.0 BASIS OF NOMINATION Ketamine, a noncompetitive NMDA receptor blocker, has been used extensively off - label as a pediatric anesthetic for surgical procedures in infants and toddlers. Recently, Olney and coworkers have demonstrated severe widespread apoptotic degeneration throughout the rapidly developing brain of the 7-day-old rat after ketamine administration. Recent research at FDA has confirmed and extended Olney’s observations. These findings are cause for concern with respect to ketamine use in children. The issue of whether the neurotoxicity found in this animal model (rat) has scientific and regulatory relevance for the pediatric use of ketamine relies heavily upon confirmation of these findings that may be obtained from the conduct of an appropriate study in non-human primates. 2.0 BACKGROUND INFORMATION The issue of potential ketamine neurotoxicity in children surfaced as a result of FDA’s reluctance to approve an NIH pediatric clinical trial using this compound because of its documented neurotoxic effects in young rats (published in several papers over the last ten years by Olney and co-workers). -

The Anticholinergic Toxidrome
Poison HOTLINE Partnership between Iowa Health System and University of Iowa Hospitals and Clinics July 2011 The Anticholinergic Toxidrome A toxidrome is a group of symptoms associated with poisoning by a particular class of agents. One example is the opiate toxidrome, the triad of CNS depression, respiratory depression, and pinpoint pupils, and which usually responds to naloxone. The anticholinergic toxidrome is most frequently associated with overdoses of diphenhydramine, a very common OTC medication. However, many drugs and plants can produce the anticholinergic toxidrome. A partial list includes: tricyclic antidepressants (amitriptyline), older antihistamines (chlorpheniramine), Did you know …… phenothiazines (promethazine) and plants containing the anticholinergic alkaloids atropine, hyoscyamine and scopolamine (Jimson Weed). Each summer, the ISPCC receives approximately 10-20 The mnemonic used to help remember the symptoms and signs of this snake bite calls, some being toxidrome are derived from the Alice in Wonderland story: from poisonous snakes (both Blind as a Bat (mydriasis and inability to focus on near objects) local and exotic). Red as a Beet (flushed skin color) Four poisonous snakes can be Hot as Hades (elevated temperature) found in Iowa: the prairie These patients can sometimes die of agitation-induced hyperthermia. rattlesnake, the massasauga, Dry as a Bone (dry mouth and dry skin) the copperhead, and the Mad as a Hatter (hallucinations and delirium) timber rattlesnake. Each Bowel and bladder lose their tone (urinary retention and constipation) snake has specific territories Heart races on alone (tachycardia) within the state. ISPCC A patient who has ingested only an anticholinergic substance and is not specialists have access to tachycardic argues against a serious anticholinergic overdose. -

3 Drugs That May Cause Delirium Or Problem Behaviors CARD 03 19 12 JUSTIFIED.Pub
Drugs that May Cause Delirium or Problem Behaviors Drugs that May Cause Delirium or Problem Behaviors This reference card lists common and especially problemac drugs that may Ancholinergics—all drugs on this side of the card. May impair cognion cause delirium or contribute to problem behaviors in people with demena. and cause psychosis. Drugs available over‐the‐counter marked with * This does not always mean the drugs should not be used, and not all such drugs are listed. If a paent develops delirium or has new problem Tricyclic Andepressants Bladder Anspasmodics behaviors, a careful review of all medicaons is recommended. Amitriptyline – Elavil Darifenacin – Enablex Be especially mindful of new medicaons. Clomipramine – Anafranil Flavoxate – Urispas Desipramine – Norpramin Anconvulsants Psychiatric Oxybutynin – Ditropan Doxepin – Sinequan Solifenacin – VESIcare All can cause delirium, e.g. All psychiatric medicaons should be Imipramine – Tofranil Tolterodine – Detrol Carbamazepine – Tegretol reviewed as possible causes, as Nortriptyline – Aventyl, Pamelor Gabapenn – Neuronn effects are unpredictable. Trospium – Sanctura Anhistamines / Allergy / Leveracetam – Keppra Notable offenders include: Insomnia / Sleep Valproic acid – Depakote Benzodiazepines e.g. Cough & Cold Medicines *Diphenhydramine – Sominex, ‐Alprazolam – Xanax *Azelasne – Astepro Pain Tylenol‐PM, others ‐Clonazepam – Klonopin *Brompheniramine – Bromax, All opiates can cause delirium if dose *Doxylamine – Unisom, Medi‐Sleep ‐Lorazepam – Avan Bromfed, Lodrane is too high or -

(12) United States Patent (10) Patent No.: US 8,486,621 B2 Luo Et Al
USOO8486.621B2 (12) United States Patent (10) Patent No.: US 8,486,621 B2 Luo et al. (45) Date of Patent: Jul. 16, 2013 (54) NUCLEICACID-BASED MATRIXES 2005. O130180 A1 6/2005 Luo et al. 2006/0084607 A1 4/2006 Spirio et al. (75) Inventors: ity, N. (US); Soong Ho 2007/01482462007/0048759 A1 3/20076/2007 Luo et al. m, Ithaca, NY (US) 2008.0167454 A1 7, 2008 Luo et al. 2010, O136614 A1 6, 2010 Luo et al. (73) Assignee: Cornell Research Foundation, Inc., 2012/0022244 A1 1, 2012 Yin Ithaca, NY (US) FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this WO WO 2004/057023 A1 T 2004 patent is extended or adjusted under 35 U.S.C. 154(b) by 808 days. OTHER PUBLICATIONS Lin et al. (J Biomech Eng. Feb. 2004;126(1):104-10).* (21) Appl. No.: 11/464,184 Li et al. (Nat Mater. Jan. 2004:3(1):38-42. Epub Dec. 21, 2003).* Ma et al. (Nucleic Acids Res. Dec. 22, 1986;14(24):9745-53).* (22) Filed: Aug. 11, 2006 Matsuura, et al. Nucleo-nanocages: designed ternary oligodeoxyribonucleotides spontaneously form nanosized DNA (65) Prior Publication Data cages. Chem Commun (Camb). 2003; (3):376-7. Li, et al. Multiplexed detection of pathogen DNA with DNA-based US 2007/01 17177 A1 May 24, 2007 fluorescence nanobarcodes. Nat Biotechnol. 2005; 23(7): 885-9. Lund, et al. Self-assembling a molecular pegboard. JAm ChemSoc. Related U.S. Application Data 2005; 127(50): 17606-7. (60) Provisional application No. 60/722,032, filed on Sep. -

Medicare Part D Excluded Drug List
MEDICARE PART D EXCLUDED DRUGS LIST 2016_updated July 2016 Reason: LIST = multiple reasons it's excluded; "not covered under Part D law" Reason: Not properly listed with FDA = CMS considers it best practice for Part D sponsors to consider the proper listing of a drug product with the FDA as a prerequisite for making a Part D drug coverage determination. The FDA is unable to provide regulatory status determinations through their regular processes if a drug product is not properly listed. Therefore, Part D sponsors should begin the drug coverage determination process by confirming that a prescription drug product national drug code (NDC) is properly listed with the FDA. Reason: DESI = Less Than Effective (LTE) drug for ALL indications Label Name Reason 1ST BASE CRE Bulk Ingredient 2-FUCCOSYLLA PAK LACTO-N Medical Food ABANEU-SL SUB Vitamin/Mineral ABLAVAR INJ 244MG/ML Diagnostic Agent ACACIA EXTRA SOL 1:20 Non-standardized allergenic ACCUCAINE INJ 1% LIST ACD FORMULA SOL A Blood Component ACD-A SOL Blood Component ACLARO PD EMU 4% Cosmetic ACREMONIUM SOL 20000PNU Non-standardized allergenic ACTCT FLEX 3 PAD 4"X4" Not properly listed with FDA ACTHREL INJ 100MCG Diagnostic Agent ACTI ANTIMIC PAD 2"X2" Not properly listed with FDA ACTI ANTIMIC PAD 4"X4" Not properly listed with FDA ACTICOAT 7 PAD 2"X2" Not properly listed with FDA ACTICOAT 7 PAD 4"X5" Not properly listed with FDA ACTICOAT ABS PAD 4"X5" Not properly listed with FDA ACTICOAT MOI PAD 2"X2" Surgical Supply/Medical ACTICOAT MOI PAD 4"X4" Surgical Supply/Medical ACTICOAT MOI PAD -

(12) United States Patent (10) Patent N0.: US 7,964,607 B2 Verhoest Et A1
US007964607B2 (12) United States Patent (10) Patent N0.: US 7,964,607 B2 Verhoest et a1. (45) Date of Patent: Jun. 21, 2011 (54) PYRAZOLO[3,4-D]PYRIMIDINE FOREIGN PATENT DOCUMENTS COMPOUNDS EP 1460077 9/2004 WO 02085904 10/2002 (75) Inventors: Patrick Robert Verhoest, Old Lyme, CT WO 2004037176 5/2004 (US); Caroline ProulX-Lafrance, Ledyard, CT (US) OTHER PUBLICATIONS Wunder et a1, M01. PharmacoL, v01. 28, N0. 6, (2005), pp. 1776 (73) Assignee: P?zer Inc., New York, NY (U S) 1781. van der Staay et a1, Neuropharmacology, v01. 55 (2008), pp. 908 ( * ) Notice: Subject to any disclaimer, the term of this 918. patent is extended or adjusted under 35 USC 154(b) by 562 days. Primary Examiner * Susanna Moore (74) Attorney, Agent, or Firm * Jennifer A. Kispert; (21) Appl.No.: 12/118,062 Michael Herman (22) Filed: May 9, 2008 (57) ABSTRACT (65) Prior Publication Data The invention provides PDE9-inhibiting compounds of For US 2009/0030003 A1 Jan. 29, 2009 mula (I), Related US. Application Data (60) Provisional application No. 60/917,333, ?led on May 11, 2007. (51) Int. Cl. C07D 48 7/04 (2006.01) A61K 31/519 (2006.01) A61P 25/28 (2006.01) (52) US. Cl. ................................... .. 514/262.1; 544/262 (58) Field of Classi?cation Search ................ .. 544/262; 5 1 4/2 62 .1 See application ?le for complete search history. and pharmaceutically acceptable salts thereof, Wherein R, R1, (56) References Cited R2 and R3 are as de?ned herein. Pharmaceutical compositions containing the compounds of Formula I, and uses thereof in U.S. -

PR2'2006.Vp:Corelventura
Modulation of chemical signalling: Current problems after four decades of research Symposium 5-HT3 receptors and emesis Martin Barann1,2, Michael Bruess2, Marion Brinkmann1, Isabelle Linden1, Mina Lyutenska1, Marc Schneider1, Jan Walkembach1, Maria Wittmann1 Clinics of Anaesthesiology and Operative Intensive Care, University of Bonn, Sigmund-Freud-Str. 25, D-53105 Bonn, Germany; Institute of Pharmacology and Toxicology, University of Bonn, Reuterstr. 2b, D-53113 Bonn, Germany; e-mail: [email protected] Nausea and vomiting. Nausea and vomiting is a ma- mechanisms. In recent studies, the molecular mecha- jor problem which occurs during and after the treat- nisms of (anti)emetic drugs at this receptor have been ment with certain drugs like anesthetics, opioid- studied [Barann, et al., Naunyn Schmiedebergs Arch analgesics or cytostatics. Postoperative nausea and Pharmacol, 2000; Barann et al., Neuropharmacology, vomiting (PONV) is called the “big” little problem. 2000; Barann et al., Br J Pharmacol, 2002; Walkem- As a consequence, the choice of the anesthetic drug(s) bach et al., Br J Pharmacol, 2005; Barann et al., Eur J plays a role in preventing PONV [Apfel et al., Anes- Pharamacol, 2006]. For this purpose, excised thesiology, 1999; Eberhart et al., Eur J Anaesthesiol, outside-out patches of HEK293 cells, stably trans- via 1999]. Nausea and vomiting can be mediated pe- fected with the human 5-HT3A receptor cDNA were ripheral and/or central nervous pathways. Within the formed (voltage-clamp mode) and fast solution exchange peripheral nervous system, vagal afferents, which are systems were used. In addition, radioligand binding stud- stimulated by 5-HT released from enterochromaffin ies and [3H]5-HT uptake measurements (study of the cells, are of importance. -

Biol. Pharm. Bull. 42(5): 736-743 (2019)
736 Biol. Pharm. Bull. 42, 736–743 (2019) Vol. 42, No. 5 Regular Article Noradrenaline-Induced Relaxation of Urinary Bladder Smooth Muscle Is Primarily Triggered through the β3-Adrenoceptor in Rats Keisuke Obara,a Serena Suzuki,a Hiroko Shibata,a Naoki Yoneyama, a Shoko Hamamatsu,a Fumiko Yamaki,a Koji Higai,b and Yoshio Tanaka*,a a Department of Chemical Pharmacology, Faculty of Pharmaceutical Sciences, Toho University; 2–2–1 Miyama, Funabashi, Chiba 274–8510, Japan: and b Laboratory of Medical Biochemistry, Faculty of Pharmaceutical Sciences, Toho University; 2–2–1 Miyama, Funabashi, Chiba 274–8510, Japan. Received November 18, 2018; accepted January 25, 2019 β-Adrenoceptors are subclassified into 3 subtypes (β1–β3). Among these, β3-adrenoceptors are present in various types of smooth muscle and are believed to play a role in relaxation responses of these muscles. β3-Adrenoceptors are also present in urinary bladder smooth muscle (UBSM), although their expression varies depending on the animal species. To date, there has been little information available about the endo- genous ligand that stimulates β3-adrenoceptors to produce relaxation responses in UBSM. In this study, to determine whether noradrenaline is a ligand of UBSM β3-adrenoceptors, noradrenaline-induced relaxation was analyzed pharmacologically using rat UBSM. We also assessed whether noradrenaline metabolites were ligands in UBSM. In isolated rat urinary bladder tissues, mRNAs for β1-, β2-, and β3-adrenoceptors were detected using RT-PCR. In UBSM preparations contracted with methacholine (3 105 M), noradrenaline- 6 induced relaxation was not inhibited by the following antagonists: atenolol (10 M; selective β1-adrenoceptor 8 7 antagonist), ICI-118,551 (3 10 M; selective β2-adrenoceptor antagonist), propranolol (10 M; non-selective β-adrenoceptor antagonist), and bupranolol (107 M; non-selective β-adrenoceptor antagonist). -

Hyoscyamine, Atropine, Scopolamine, and Phenobarbital
PATIENT & CAREGIVER EDUCATION Hyoscyamine, Atropine, Scopolamine, and Phenobarbital This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Donnatal; Phenohytro What is this drug used for? It is used to treat irritable bowel syndrome. It is used to treat ulcer disease. It may be given to your child for other reasons. Talk with the doctor. What do I need to tell the doctor BEFORE my child takes this drug? If your child is allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell the doctor about the allergy and what signs your child had. If your child has any of these health problems: Heart problems due to bleeding, bowel block, enlarged colon, glaucoma, a hernia Hyoscyamine, Atropine, Scopolamine, and Phenobarbital 1/9 that involves your stomach (hiatal hernia), myasthenia gravis, slow-moving GI (gastrointestinal) tract, trouble passing urine, or ulcerative colitis. If your child has ever had porphyria. If your child has been restless or overexcited in the past after taking phenobarbital. If your child is addicted to drugs or alcohol, or has been in the past. This is not a list of all drugs or health problems that interact with this drug. Tell the doctor and pharmacist about all of your child’s drugs (prescription or OTC, natural products, vitamins) and health problems. You must check to make sure that it is safe to give this drug with all of your child’s other drugs and health problems. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.