Chlorodifluoromethane Hydrodechlorination On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effect of Insulations on Cop in Vapor Compression Refrigeration System
International Journal of Mechanical Engineering and Technology (IJMET) Volume 10, Issue 01, January 2019, pp. 1201-1208, Article ID: IJMET_10_01_122 Available online at http://www.iaeme.com/ijmet/issues.asp?JType=IJMET&VType=10&IType= 01 ISSN Print: 0976-6340 and ISSN Online: 0976-6359 © IAEME Publication Scopus Indexed EFFECT OF INSULATIONS ON COP IN VAPOR COMPRESSION REFRIGERATION SYSTEM Anusha Peyyala Assistant Professor P V P Siddhartha Institute of Technology, Research Scholar, Acharya Nagarjuna University, India. Dr N V V S Sudheer Associate Professor, R V R & J C College of Engineering, Guntur India ABSTRACT In this project, experimentation is done on Vapour Compression Refrigeration System [VCRS] as the COP is high for this system and it is the present trend of the HVAC in the domestic industry. This study presents investigation of best suited refrigerant and insulation combination for gas pipeline and liquid pipeline of a split air conditioning system. Analysis are performed for R22-Chlorodiflouromethane, a HydroChloroFlouro Carbon refrigerant, which has been using in the present world that cause both global warming and ozone layer depletion and R410a, mixture of di- flouromethane and pentaflouroethane, a Hydroflouro carbon refrigerant, which is future of HVAC which reduces the effect of ozone layer depletion [ODP] and Global Warming Potential [GWP].For these two refrigerants, we had found out the best insulation suitable as insulation also affects the COP of air conditioner, which has been observed from the literature. Minimizing the temperature of refrigerant in suction line helps condensing unit work more effectively intern the system performance increases. This reduces the overall power required for working of air conditioner, thereby reducing the maintenance cost of system. -

New and Improved Infrared Absorption Cross Sections for Chlorodifluoromethane (HCFC-22)
Atmos. Meas. Tech., 9, 2593–2601, 2016 www.atmos-meas-tech.net/9/2593/2016/ doi:10.5194/amt-9-2593-2016 © Author(s) 2016. CC Attribution 3.0 License. New and improved infrared absorption cross sections for chlorodifluoromethane (HCFC-22) Jeremy J. Harrison1,2 1Department of Physics and Astronomy, University of Leicester, University Road, Leicester, LE1 7RH, UK 2National Centre for Earth Observation, University of Leicester, University Road, Leicester, LE1 7RH, UK Correspondence to: Jeremy J. Harrison ([email protected]) Received: 10 December 2015 – Published in Atmos. Meas. Tech. Discuss.: 18 January 2016 Revised: 3 May 2016 – Accepted: 6 May 2016 – Published: 17 June 2016 Abstract. The most widely used hydrochlorofluorocarbon 1 Introduction (HCFC) commercially since the 1930s has been chloro- difluoromethane, or HCFC-22, which has the undesirable The consumer appetite for safe household refrigeration led effect of depleting stratospheric ozone. As this molecule to the commercialisation in the 1930s of dichlorodifluo- is currently being phased out under the Montreal Pro- romethane, or CFC-12, a non-flammable and non-toxic re- tocol, monitoring its concentration profiles using infrared frigerant (Myers, 2007). Within the next few decades, other sounders crucially requires accurate laboratory spectroscopic chemically related refrigerants were additionally commer- data. This work describes new high-resolution infrared ab- cialised, including chlorodifluoromethane, or a hydrochlo- sorption cross sections of chlorodifluoromethane over the rofluorocarbon known as HCFC-22, which found use in a spectral range 730–1380 cm−1, determined from spectra wide array of applications such as air conditioners, chillers, recorded using a high-resolution Fourier transform spectrom- and refrigeration for food retail and industrial processes. -

SAFETY DATA SHEET Chlorodifluoromethane (R 22) Issue Date: 16.01.2013 Version: 1.0 SDS No.: 000010021746 Last Revised Date: 18.06.2015 1/15
SAFETY DATA SHEET Chlorodifluoromethane (R 22) Issue Date: 16.01.2013 Version: 1.0 SDS No.: 000010021746 Last revised date: 18.06.2015 1/15 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifier Product name: Chlorodifluoromethane (R 22) Trade name: Gasart 503 R22 Additional identification Chemical name: Chlorodifluoromethane Chemical formula: CHClF2 INDEX No. - CAS-No. 75-45-6 EC No. 200-871-9 REACH Registration No. 01-2119517587-31 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses: Industrial and professional. Perform risk assessment prior to use. Refrigerant. Using gas alone or in mixtures for the calibration of analysis equipment. Using gas as feedstock in chemical processes. Formulation of mixtures with gas in pressure receptacles. Uses advised against Consumer use. 1.3 Details of the supplier of the safety data sheet Supplier Linde Gas GmbH Telephone: +43 50 4273 Carl-von-Linde-Platz 1 A-4651 Stadl-Paura E-mail: [email protected] 1.4 Emergency telephone number: Emergency number Linde: + 43 50 4273 (during business hours), Poisoning Information Center: +43 1 406 43 43 SDS_AT - 000010021746 SAFETY DATA SHEET Chlorodifluoromethane (R 22) Issue Date: 16.01.2013 Version: 1.0 SDS No.: 000010021746 Last revised date: 18.06.2015 2/15 SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Directive 67/548/EEC or 1999/45/EC as amended. N; R59 The full text for all R-phrases is displayed in section 16. Classification according to Regulation (EC) No 1272/2008 as amended. -

UNITED STATES PATENT of FICE 2,640,086 PROCESS for SEPARATING HYDROGEN FLUORIDE from CHLORODFLUORO METHANE Robert H
Patented May 26, 1953 2,640,086 UNITED STATES PATENT of FICE 2,640,086 PROCESS FOR SEPARATING HYDROGEN FLUORIDE FROM CHLORODFLUORO METHANE Robert H. Baldwin, Chadds Ford, Pa., assignor to E. H. du Pont de Nemours and Company, Wi inington, Del, a corporation of Delaware No Drawing. Application December 15, 1951, Serial No. 261,929 9 Claims. (C. 260-653) 2 This invention relates to a process for Sep These objects are accomplished essentially by arating hydrogen fluoride from monochlorodi Subjecting a mixture of hydrogen fluoride and fluoronethane, and more particularly, separat Inonochlorodifluoromethane in the liquid phase ing these components from the reaction mixture to temperatures below 0° C., preferably at about obtained in the fluorination of chloroform with -30° C. to -50° C., at either atmospheric or hydrogen fluoride, Super-atmospheric pressures, together with from In the fluorination of chloroform in the prest about 0.25 mol to about 2.5 mols of chloroform ence Of a Catalyst, a reaction mixture is pro per mol of chlorodifluoronethane contained in duced which consists essentially of HCl, HF, the mixture and separating an upper layer rich CHCIF2, CHCl2F, CHCls, and CHF3. A method O in HF from a lower organic layer. The proceSS of Separating these components is disclosed in is operative with mixtures containing up to 77% U. S. Patent No. 2,450,414 which involves sep by weight of HF. arating the components by a special fractional It has been found that chloroform is substan distillation under appropriate temperatures and tially immiscible With EIF at temperatures be pressures. -

CYCLE D-HX: NIST Vapor Compression Cycle Model Accounting for Refrigerant Thermodynamic and Transport Properties
NIST Technical Note 1974 CYCLE_D-HX: NIST Vapor Compression Cycle Model Accounting for Refrigerant Thermodynamic and Transport Properties Version 1.0 User’s Guide J.S. Brown R. Brignoli P.A. Domanski This publication is available free of charge from: https://doi.org/10.6028/NIST.TN.1974 NIST Technical Note 1974 CYCLE_D-HX: NIST Vapor Compression Cycle Model Accounting for Refrigerant Thermodynamic and Transport Properties Version 1.0 User’s Guide J.S. Brown The Catholic University of America R. Brignoli P.A. Domanski Engineering Laboratory National Institute of Standards and Technology This publication is available free of charge from: https://doi.org/10.6028/NIST.TN.1974 December 2017 U.S. Department of Commerce Wilbur L. Ross, Jr., Secretary National Institute of Standards and Technology Walter Copan, NIST Director and Under Secretary of Commerce for Standards and Technology This software package was developed by the National Institute of Standards and Technology (NIST), is not subject to copyright protection, and is in the public domain. It can be used freely provided that any derivative works bear some notice that they are derived from it. NIST used its best efforts to provide a high-quality software package and to select modeling methods and correlations based on sound scientific judgement. However, NIST assumes neither responsibility nor liability for any damage arising out of or relating to the use of CYCLE_D-HX. The software is provided “AS IS”; NIST makes NO GUARANTIES and NO WARRANTIES OF ANY TYPE, expressed or implied, including NO WARRANTY OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE. -
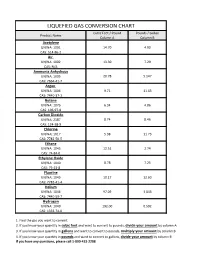
Liquefied Gas Conversion Chart
LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Acetylene UN/NA: 1001 14.70 4.90 CAS: 514-86-2 Air UN/NA: 1002 13.30 7.29 CAS: N/A Ammonia Anhydrous UN/NA: 1005 20.78 5.147 CAS: 7664-41-7 Argon UN/NA: 1006 9.71 11.63 CAS: 7440-37-1 Butane UN/NA: 1075 6.34 4.86 CAS: 106-97-8 Carbon Dioxide UN/NA: 2187 8.74 8.46 CAS: 124-38-9 Chlorine UN/NA: 1017 5.38 11.73 CAS: 7782-50-5 Ethane UN/NA: 1045 12.51 2.74 CAS: 74-84-0 Ethylene Oxide UN/NA: 1040 8.78 7.25 CAS: 75-21-8 Fluorine UN/NA: 1045 10.17 12.60 CAS: 7782-41-4 Helium UN/NA: 1046 97.09 1.043 CAS: 7440-59-7 Hydrogen UN/NA: 1049 192.00 0.592 CAS: 1333-74-0 1. Find the gas you want to convert. 2. If you know your quantity in cubic feet and want to convert to pounds, divide your amount by column A 3. If you know your quantity in gallons and want to convert to pounds, multiply your amount by column B 4. If you know your quantity in pounds and want to convert to gallons, divide your amount by column B If you have any questions, please call 1-800-433-2288 LIQUEFIED GAS CONVERSION CHART Cubic Feet / Pound Pounds / Gallon Product Name Column A Column B Hydrogen Chloride UN/NA: 1050 10.60 8.35 CAS: 7647-01-0 Krypton UN/NA: 1056 4.60 20.15 CAS: 7439-90-9 Methane UN/NA: 1971 23.61 3.55 CAS: 74-82-8 Methyl Bromide UN/NA: 1062 4.03 5.37 CAS: 74-83-9 Neon UN/NA: 1065 19.18 10.07 CAS: 7440-01-9 Mapp Gas UN/NA: 1060 9.20 4.80 CAS: N/A Nitrogen UN/NA: 1066 13.89 6.75 CAS: 7727-37-9 Nitrous Oxide UN/NA: 1070 8.73 6.45 CAS: 10024-97-2 Oxygen UN/NA: 1072 12.05 9.52 CAS: 7782-44-7 Propane UN/NA: 1075 8.45 4.22 CAS: 74-98-6 Sulfur Dioxide UN/NA: 1079 5.94 12.0 CAS: 7446-09-5 Xenon UN/NA: 2036 2.93 25.51 CAS: 7440-63-3 1. -

R-22 Safety Data Sheet
R-22 Safety Data Sheet R-22 1. CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME: R-22 OTHER NAME: Chlorodifluoromethane USE: Refrigerant Gas DISTRIBUTOR: National Refrigerants, Inc. 661 Kenyon Avenue Bridgeton, New Jersey 08302 FOR MORE INFORMATION CALL: IN CASE OF EMERGENCY CALL: (Monday-Friday, 8:00am-5:00pm) CHEMTREC: 1-800-424-9300 1-800-262-0012 2. HAZARDS IDENTIFICATION CLASSIFICATION: Gases under pressure, Liquefied Gas SIGNAL WORD: WARNING HAZARD STATEMENT: Contains gas under pressure, may explode if heated SYMBOL: Gas Cylinder PRECAUTIONARY STATEMENT: STORAGE: Protect from sunlight, store in a well ventilated place EMERGENCY OVERVIEW: Colorless, volatile liquid with ethereal and faint sweetish odor. Non-flammable material. Overexposure may cause dizziness and loss of concentration. At higher levels, CNS depression and cardiac arrhythmia may result from exposure. Vapors displace air and can cause asphyxiation in confined spaces. At higher temperatures, (>250C), decomposition products may include Hydrochloric Acid (HCI), Hydrofluoric Acid (HF) and carbonyl halides. POTENTIAL HEALTH HAZARDS SKIN: Irritation would result from a defatting action on tissue. Liquid contact could cause frostbite. EYES: Liquid contact can cause severe irritation and frostbite. Mist may irritate. INHALATION: R-22 is low in acute toxicity in animals. When oxygen levels in air are reduced to 12-14% by displacement, symptoms of asphyxiation, loss of coordination, increased pulse rate and deeper respiration will occur. At high levels, cardiac arrhythmia may occur. INGESTION: Ingestion is unlikely because of the low boiling point of the material. Should it occur, discomfort in the gastrointestinal tract from rapid evaporation of the material and consequent evolution of gas would result. -

181 Chloroform 4. Production, Import/Export, Use, And
CHLOROFORM 181 4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL Chloroform is used primarily in the production of chlorodifluoromethane (hydrochlorofluorocarbon-22 or HCFC-22) used as a refrigerant for home air conditioners or large supermarket freezers and in the production of fluoropolymers (CMR 1995). Chloroform has also been used as a solvent, a heat transfer medium in fire extinguishers, an intermediate in the preparation of dyes and pesticides, and other applications highlighted below. Its use as an anesthetic has been largely discontinued. It has limited medical uses in some dental procedures and in the administration of drugs for the treatment of some diseases. 4.1 PRODUCTION The chlorination of methane and the chlorination of methyl chloride produced by the reaction of methanol and hydrogen chloride are the two common methods for commercial chloroform production (Ahlstrom and Steele 1979; Deshon 1979). The Vulcan Materials Co., Wichita, Kansas, was documented as still using the methanol production process during the late 1980s with all other facilities in the United States at that time using the methyl chloride chlorination process (SRI 1990). One U.S. manufacturer began chloroform production in 1903, but significant commercial production was not reported until 1922 (IARC 1979). Since the early 1980s, chloroform production increased by 20-25%, due primarily to a higher demand for HCFC-22, the major chemical produced from chloroform. The Montreal Protocol established goals for phasing out the use of a variety of ozonedepleting chemicals, including most chlorofluorocarbons (CFCs). HCFC-22 was one of the few fluorocarbons not restricted by the international agreement. Chloroform is used in the manufacture of HCFC-22, and an increase in the production of this refrigerant has led to a modest increase in the demand for chloroform (CMR 1989). -

Utilization of Fluoroform for Difluoromethylation in Continuous
Green Chemistry View Article Online COMMUNICATION View Journal | View Issue Utilization of fluoroform for difluoromethylation in continuous flow: a concise synthesis of Cite this: Green Chem., 2018, 20, 108 α-difluoromethyl-amino acids† Received 26th September 2017, Accepted 30th October 2017 Manuel Köckinger,a Tanja Ciaglia,a Michael Bersier,b Paul Hanselmann,b DOI: 10.1039/c7gc02913f Bernhard Gutmann *a,c and C. Oliver Kappe *a,c rsc.li/greenchem Fluoroform (CHF3) can be considered as an ideal reagent for trolled under the Montreal Protocol. As a consequence, its pro- difluoromethylation reactions. However, due to the low reactivity duction and usage has become increasingly limited and expen- of fluoroform, only very few applications have been reported so sive. A plethora of alternative difluoromethane sources have far. Herein we report a continuous flow difluoromethylation proto- been developed in recent years, including TMSCF2Br, 3 2 col on sp carbons employing fluoroform as a reagent. The proto- (EtO)2POCF2Br, PhCOCF2Cl and CHF2OTf. Although these col is applicable for the direct Cα-difluoromethylation of protected reagents cover the needs of chemists for difluoromethylation Creative Commons Attribution 3.0 Unported Licence. α-amino acids, and enables a highly atom efficient synthesis of the on a laboratory scale, their high cost, low atom economy and active pharmaceutical ingredient eflornithine. limited commercial availability prohibit their usage in an industrial setting. The difluoromethyl group is found in an increasing array of The most attractive CF - and CHF -source is fluoroform 1 3 2 pharmaceutical and agrochemical products. Not surprisingly, (CHF , Freon 23). Fluoroform is generated as a large-volume ff 3 therefore, significant e orts have been devoted towards the waste-product during the synthesis of chlorodifluoromethane development of novel protocols for the introduction of the (Fig. -

Direct Nucleophilic Trifluoromethylation of Carbonyl Compounds By
www.nature.com/scientificreports OPEN Direct nucleophilic trifuoromethylation of carbonyl compounds by potent greenhouse Received: 16 April 2018 Accepted: 18 July 2018 gas, fuoroform: Improving Published: xx xx xxxx the reactivity of anionoid trifuoromethyl species in glymes Takuya Saito1, Jiandong Wang1, Etsuko Tokunaga1, Seiji Tsuzuki2 & Norio Shibata 1,3 A simple protocol to overcome the problematic trifuoromethylation of carbonyl compounds by the potent greenhouse gas “HFC-23, fuoroform” with a potassium base is described. Simply the use of glymes as a solvent or an additive dramatically improves the yields of this transformation. Experimental results and DFT calculations suggest that the benefcial efect deals with glyme coordination to the + + K to produce [K(polyether)n] whose diminished Lewis acidity renders the reactive anionoid CF3 − counterion species more ‘naked’, thereby slowing down its undesirable decomposition to CF2 and F and simultaneously increasing its reactivity towards the organic substrate. Tere has been remarkable progress recently in the synthetic incorporation of a trifuoromethyl (CF3) moiety into potential bioactive molecules, prompting the discovery of new pharmaceuticals and agrochemicals1–5. Fluoroform (HFC-23, HCF3, trifuoromethane) is a potent greenhouse gas that is formed as a by-product in huge amounts during the synthesis of poly-tetrafuoroethylene (PTFE) and polyvinylidene difuoride (PVDF) from chlorodifuoromethane (ClCHF2). Fluoroform has a 11,700-fold higher GWP than carbon dioxide with an atmos- pheric lifetime of 264 years and is used to a very limited extent as a refrigerant or as a raw material6–10. At present, fuoroform abatement techniques involve thermal oxidation, catalytic hydrolysis and plasma destruction, so there are operation and economical limits to transform fuoroform to useful refrigerants or fre extinguishers11–17. -

Honeywell International Inc
SAFETY DATA SHEET Genetron® HP80 (R-402A) 000000009892 Version 2.6 Revision Date 04/10/2014 Print Date 06/22/2015 SECTION 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Genetron® HP80 (R-402A) MSDS Number : 000000009892 Product Use Description : Refrigerant Manufacturer or supplier's : Honeywell International Inc. details 101 Columbia Road Morristown, NJ 07962-1057 For more information call : 800-522-8001 +1-973-455-6300 (Monday-Friday, 9:00am-5:00pm) In case of emergency call : Medical: 1-800-498-5701 or +1-303-389-1414 : Transportation (CHEMTREC): 1-800-424-9300 or +1-703-527-3887 : : (24 hours/day, 7 days/week) SECTION 2. HAZARDS IDENTIFICATION Emergency Overview Form : Liquefied gas Color : colourless Odor : weak Classification of the substance or mixture Classification of the substance : Gases under pressure, Liquefied gas or mixture Simple Asphyxiant GHS Label elements, including precautionary statements Page 1 / 16 SAFETY DATA SHEET Genetron® HP80 (R-402A) 000000009892 Version 2.6 Revision Date 04/10/2014 Print Date 06/22/2015 Symbol(s) : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. Precautionary statements : Prevention: Use personal protective equipment as required. Storage: Protect from sunlight. Store in a well-ventilated place. Hazards not otherwise : May cause eye and skin irritation. classified May cause frostbite. May cause cardiac arrhythmia. Carcinogenicity No component of this product present at levels greater than or equal to 0.1% is identified as a known or anticipated carcinogen by NTP, IARC, or OSHA. SECTION 3. COMPOSITION/INFORMATION ON INGREDIENTS Chemical nature : Mixture Chemical Name CAS-No. -

Phase Behavior of Water-Insoluble Simvastatin Drug in Supercritical Mixtures of Chlorodifluoromethane and Carbon Dioxide
Korean J. Chem. Eng., 23(6), 1009-1015 (2006) SHORT COMMUNICATION Phase behavior of water-insoluble simvastatin drug in supercritical mixtures of chlorodifluoromethane and carbon dioxide Dong-Joon Oh, Byung-Chul Lee† and Sung-Joo Hwang* Department of Chemical Engineering and Nano-Bio Technology, Hannam University, 133 Ojung-dong, Daeduk-gu, Daejeon 306-791, Korea *College of Pharmacy, Chungnam National University, 220 Kung-dong, Yusong-gu, Daejeon 305-764, Korea (Received 1 May 2006 • accepted 16 May 2006) Abstract−Phase behavior data are presented for simvastatin, a water-insoluble drug, in supercritical solvent mixtures of chlorodifluoromethane (CHClF2) and carbon dioxide (CO2). The solubilities of the simvastatin drug in the solvent mixtures of CHClF2 and CO2 were determined by measuring the cloud point pressures using a variable-volume view cell apparatus as functions of temperature, solvent composition, and amount of the drug loaded into the solution. The cloud point pressure increased with increasing the system temperature. As the CHClF2 composition in the solvent mix- ture increased, the cloud point pressure at a fixed temperature decreased. Addition of CHClF2 to CO2 caused an increase of the dissolving power of the mixed solvent for the simvastatin drug due to the increase of the solvent polarity. CHClF2 acted as a solvent for simvastatin, while CO2 acted as an anti-solvent. The cloud point pressure increased as the amount of the simvastatin drug in the solvent mixture increased. Consequently, the solubility of the simvastatin drug in the solvent mixture of CHClF2 and CO2 decreased with increasing the CO2 content in the solvent mixture as well as with increasing the system temperature.