Implications for Their Dietary Essentiality and Use As Supplements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fatty Acids: Essential…Therapeutic
Volume 3, No.2 May/June 2000 A CONCISE UPDATE OF IMPORTANT ISSUES CONCERNING NATURAL HEALTH INGREDIENTS Written and Edited By: Thomas G. Guilliams Ph.D. FATTY ACIDS: Essential...Therapeutic Few things have been as confusing to both patient and health care provider as the issue of fats and oils. Of all the essential nutrients required for optimal health, fatty acids have not only been forgotten they have been considered hazardous. Health has somehow been equated with “low-fat” or “fat-free” for so long, to suggest that fats could be essential or even therapeutic is to risk credibility. We hope to give a view of fats that is both balanced and scientific. This review will cover the basics of most fats that will be encountered in dietary or supplemental protocols. Recommendations to view essential fatty acids in a similar fashion as essential vitamins and minerals will be combined with therapeutic protocols for conditions ranging from cardiovascular disease, skin conditions, diabetes, nerve related disorders, retinal disorders and more. A complete restoration of health cannot be accomplished until there is a restoration of fatty acid nutritional information among health care professionals and their patients. Fats- What are they? Dietary fats come to us from a variety of sources, but primarily in the form of triglycerides. That is, three fatty acid molecules connected by a glycerol backbone (see fatty acid primer page 3 for diagram). These fatty acids are then used as energy by our cells or modified into phospholipids to be used as cell or organelle membranes. Some fatty acids are used in lipoprotein molecules to shuttle cholesterol and fats to and from cells, and fats may also be stored for later use. -

Production of 8,11,14,17-Cis-Eicosatetraenoic Acid
Production of 8,11,14,17-cis-Eicosatetraenoic Acid (20:4ω-3) by a ∆5 and ∆12 Desaturase-Defective Mutant of an Arachidonic Acid-Producing Fungus Mortierella alpina 1S-4 Hiroshi Kawashimaa, Eiji Sakuradanib, Nozomu Kamadab, Kengo Akimotoa, Kyoko Konishia, Jun Ogawab, and Sakayu Shimizub,* aInstitute for Biomedical Research, Suntory Ltd., Shimamoto-cho, Osaka 618-0001, Japan, and bDivision of Applied Life Sci- ences, Graduate School of Agriculture, Kyoto University, Kyoto 606-8502, Japan ∆ ∆ ABSTRACT: A 5 and 12 desaturase-defective mutant of an there are large amounts of other C20 PUFA, such as AA, EPA, arachidonic acid-producing fungus, Mortierella alpina 1S-4, pro- and dihomo-γ-linolenic acid (DGLA), which are difficult to duced 8,11,14,17-cis-eicosatetraenoic acid (20:4ω3) intracellu- separate from 20:4ω3. During our studies on PUFA production larly when grown with linseed oil. Dihomo-γ-linolenic acid was involving AA-producing Mortierella fungi, we found 20:4ω3 the only C20 polyunsaturated fatty acid (4.9 wt% of total mycelial was formed (ca. 0.5 mg/mL of culture medium) as a by-prod- ω fatty acids) other than 20:4 3. AA and 5,8,11,14,17-cis-eicosapen- uct (3). Large amounts of AA (2.44 mg/mL) and EPA (1.88 taenoic acid were not detected. The mycelial lipids consisted of mg/mL) were produced in this case. Recently, we reported 82.2% (by mol) triacylglycerol (TG), 7.1% diacylglycerol, 8.9% ω ∆ phospholipids (PL), and 1.9% free fatty acids. The percentage of 20:4 3 production by cultivating 5 desaturase-defective mu- 20:4ω3 was higher in PL (30.1%) than in TG (11.6%), and highest tants of AA-producing M. -

Omega 3 Fatty Acids and Health
Dietary n-6 and n-3 fatty acids: health and disease Professor Asim K. Duttaroy Department of Nutrition Room No. 2199 Tel: 22 85 15 47 Email: [email protected] Type of Dietary Fatty Acids Saturated fatty acids (Palmitic acid, 16:0, Stearic acid, 18:0) Monounsaturated fatty acids (Palmiteloic acid, 16:1,n-7, Oleic acid, 18:1n-9) Polyunsaturated fatty acids (Linoleic acid, 18:2,n-6, Alpha linolenic acid, 18:3n-3) Essential Fatty acids (EFA) Long chain polyunsaturated fatty acids Arachidonic acid, 20:4n-6, Eicosapentaenoic acid, 20:5n-3, Docosahexaenoic acid, 22:6n-3 Essential Fatty acids Linoleic acid, 18:2n-6 COOH Alpha linolenic acid, 18:3n-3 COOH Metabolic Conversions of Essential Fatty Acids in Mammals N6-Series N3-Series Linoleic (18:2) α-linolenic (18:3) Δ6-desaturase γ-linolenic (18:3) Octadecatetraenoic (18:4) Elongase Dihomo−γ−linolenic (20:3) Eicosatetraenoic (20:4) Δ5-desaturase Arachidonic (20:4) Eicosapentaenoic (20:5) Elongase Adrenic (22:4) Docosapentaenoic (22:5) Elongase Tetracosatetraenoic (24:4) Tetracosapentaenoic (24:5) Δ6-desaturase Tetracosapentaenoic (24:5) Tetracosahexaenoic (24:6) β-oxidation Docosapentaenoic (22:5) (Peroxisome) Docosahexaenoic (22:6) Contributions of dietary fats to PUFA Metabolism Seed oils Sunflower, N-6 fatty acids N-6 fatty acids Flaxseed oils Safflower, Peanut Linoleic acid 18:2 α Linolenic acid, 18:3 Green leaves Evening Octadecatetraenoic acid, 18:4 Primrose, γ Linolenic acid 18:3 Borage oil Eicosatetraenoic acid,20:4 Fish, Dihomo γ Linolenic acid 20:3 Egg Eicosapentaenoic acid,20:5 -

A Role for the Orphan Human Cytochrome P450 2S1 in Polyunsaturated Fatty Acid V-1 Hydroxylation Using an Untargeted Metabolomic Approach S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2019/09/11/dmd.119.089086.DC1 1521-009X/47/11/1325–1332$35.00 https://doi.org/10.1124/dmd.119.089086 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 47:1325–1332, November 2019 Copyright ª 2019 by The American Society for Pharmacology and Experimental Therapeutics A Role for the Orphan Human Cytochrome P450 2S1 in Polyunsaturated Fatty Acid v-1 Hydroxylation Using an Untargeted Metabolomic Approach s Mostafa I. Fekry, Yi Xiao,1 Jeannette Zinggeler Berg,2 and F. Peter Guengerich Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee (M.I.F., Y.X., J.Z.B., F.P.G.); and Department of Pharmacognosy, Faculty of Pharmacy, Cairo University, Cairo, Egypt (M.I.F.) Received August 16, 2019; accepted September 6, 2019 ABSTRACT Cytochrome P450 (P450) 2S1 is one of the orphan P450s, known to of hydroxylation were slow, but no detectable activity was seen with be expressed but not having a defined function with an endogenous either medium-chain length or saturated fatty acids. P450 2S1 is Downloaded from substrate or in drug oxidations. Although it has been clearly known to be expressed, at least at the mRNA level, to the extent of demonstrated to catalyze reductive reactions, its role in NADPH- some other non-3A subfamily P450s in the human gastrointestinal dependent oxidations has been ambiguous. In our efforts to tract, and the activity may be relevant. We conclude that P450 2S1 is characterize orphan human P450 enzymes, we used an untargeted afattyacidv-1 hydroxylase, although the physiologic relevance of liquid chromatography-mass spectromterymetabolomic approach these oxidations remains to be established. -

Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty
nutrients Review Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty Acids on Human Health: An Update for 2021 Ivana Djuricic 1 and Philip C. Calder 2,3,* 1 Department of Bromatology, Faculty of Pharmacy, University of Belgrade, 11221 Belgrade, Serbia; [email protected] 2 School of Human Development and Health, Faculty of Medicine, University of Southampton, Southampton SO16 6YD, UK 3 NIHR Southampton Biomedical Research Centre, University Hospital Southampton NHS Foundation Trust and University of Southampton, Southampton SO16 6YD, UK * Correspondence: [email protected] Abstract: Oxidative stress and inflammation have been recognized as important contributors to the risk of chronic non-communicable diseases. Polyunsaturated fatty acids (PUFAs) may regulate the antioxidant signaling pathway and modulate inflammatory processes. They also influence hepatic lipid metabolism and physiological responses of other organs, including the heart. Longitudinal prospective cohort studies demonstrate that there is an association between moderate intake of the omega-6 PUFA linoleic acid and lower risk of cardiovascular diseases (CVDs), most likely as a result of lower blood cholesterol concentration. Current evidence suggests that increasing intake of arachidonic acid (up to 1500 mg/day) has no adverse effect on platelet aggregation and blood clotting, immune function and markers of inflammation, but may benefit muscle and cognitive performance. Many studies show that higher intakes of omega-3 PUFAs, especially eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are associated with a lower incidence of chronic diseases characterized Citation: Djuricic, I.; Calder, P.C. by elevated inflammation, including CVDs. This is because of the multiple molecular and cellular Beneficial Outcomes of Omega-6 and actions of EPA and DHA. -

Omega-3 Versus Omega-6 Polyunsaturated Fatty Acids in the Prevention and Treatment of Inflammatory Skin Diseases
International Journal of Molecular Sciences Review Omega-3 Versus Omega-6 Polyunsaturated Fatty Acids in the Prevention and Treatment of Inflammatory Skin Diseases Anamaria Bali´c 1 , Domagoj Vlaši´c 2, Kristina Žužul 3, Branka Marinovi´c 1 and Zrinka Bukvi´cMokos 1,* 1 Department of Dermatology and Venereology, University Hospital Centre Zagreb, School of Medicine University of Zagreb, Šalata 4, 10 000 Zagreb, Croatia; [email protected] (A.B.); [email protected] (B.M.) 2 Department of Ophtalmology and Optometry, General Hospital Dubrovnik, Ulica dr. Roka Mišeti´ca2, 20000 Dubrovnik, Croatia; [email protected] 3 School of Medicine, University of Zagreb, Šalata 3, 10000 Zagreb, Croatia; [email protected] * Correspondence: [email protected] Received: 31 December 2019; Accepted: 21 January 2020; Published: 23 January 2020 Abstract: Omega-3 (!-3) and omega-6 (!-6) polyunsaturated fatty acids (PUFAs) are nowadays desirable components of oils with special dietary and functional properties. Their therapeutic and health-promoting effects have already been established in various chronic inflammatory and autoimmune diseases through various mechanisms, including modifications in cell membrane lipid composition, gene expression, cellular metabolism, and signal transduction. The application of !-3 and !-6 PUFAs in most common skin diseases has been examined in numerous studies, but their results and conclusions were mostly opposing and inconclusive. It seems that combined !-6, gamma-linolenic acid (GLA), and -
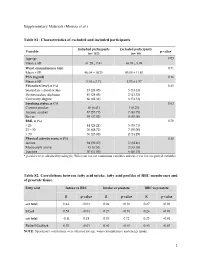
(Moussa Et Al.) Table S1
Supplementary Materials (Moussa et al.) Table S1: Characteristics of excluded and included participants Included participants Excluded participants Variable p-value* (n= 157) (n= 19) Age (y) 0.03 Mean ± SD 61.29 ± 7.41 65.05 ± 5.99 Waist circumference (cm) 0.71 Mean ± SD 96.04 ± 10.25 95.00 ± 11.83 PSA (ng/ml) 0.16 Mean ± SD 5.04 ± 2.71 4.05 ± 1.97 Education level, n (%) 0.43 Secondary school or less 45 (28.85) 5 (33.33) Postsecondary diploma 45 (28.85) 2 (13.33) University degree 66 (42.31) 8 (53.33) Smoking status, n (%) 0.63 Current smoker 10 (6.41) 1 (6.25) Former smoker 87 (55.77) 7 (43.75) Never 59 (37.82) 8 (50.00) BMI, n (%) 0.70 < 25 44 (28.21) 5 (35.71) 25 – 30 76 (48.72) 7 (50.00) > 30 36 (23.08) 2 (14.29) Physical activity score, n (%) 0.30 Active 94 (59.87) 5 (38.46) Moderately active 13 (8.28) 2 (15.38) Inactive 50 (31.85) 6 (46.15) * p-values were obtained by using the Wilcoxon test for continuous variables and chi-2 test for categorical variables. Table S2. Correlations between fatty acid intake, fatty acid profiles of RBC membranes and of prostate tissue. Fatty acid Intake vs RBC Intake vs prostate RBC vs prostate R p-value R p-value R p-value ω3 total 0.44 <0.01 0.24 <0.01 0.27 <0.01 LCω3 0.59 <0.01 0.27 <0.01 0.26 <0.01 ω6 total -0.11 0.18 0.03 0.72 0.23 <0.01 Ratio LCω3/ω6 0.53 <0.01 0.40 <0.01 0.45 <0.01 NOTE: Spearman's correlations were adjusted for age, waist circumference and energy intake. -

United States Patent (19) 11 Patent Number: 4,666,701 Horrobin Et Al
United States Patent (19) 11 Patent Number: 4,666,701 Horrobin et al. 45) Date of Patent: May 19, 1987 54 PHARMACEUTICAL AND DIETARY 58) Field of Search .......................................... 424/10. COMPOSITIONS Primary Examiner-Stanley J. Friedman 75) Inventors: David F. Horrobin, Surrey, England; Attorney, Agent, or Firm-Nixon & Vanderhye Mehar S. Manku; Yung S. Huang, both of Nova Scotia, Canada 57) ABSTRACT Efamol Limited, Surrey, England Gamma-linolenic acid or dihomo-gamma-linolenic acid 73) Assignee: for use in the reduction or prevention of gastro-intesti (21) Appl. No.: 839,228 nal bleeding and other side effects shown by NSAIDs 22 Filed: Mar, 13, 1986 when administered on a continuing basis, including use in allowing said administration to be replaced by admin (30) Foreign Application Priority Data istration of said acid alone in arthritis and other condi Mar. 19, 1985 GB United Kingdom ................. 8507058 tions without exacerbation of symptoms. 51) Int. Cl. .............................................. A61K27/00 52 U.S. C. ...................................................... 424/10 5 Claims, No Drawings 4,666,701 1. 2 been found that the 22:4n-6 acid produced from arachi PHARMACEUTICAL AND DIETARY donic acid gives rise to a series of homo-2-series PGs, COMPOSITIONS though their importance is as yet unknown. DGLA is the key substance. GLA is almost com FIELD OF THE INVENTION pletely and very rapidly converted in the body to The invention relates in one aspect to the prevention DGLA and so for practical purposes the oral adminis or reduction of side effects of aspirin and other non tration of DGLA and GLA amounts to the same thing. -

Importance of the Role of -3 and -6 Polyunsaturated Fatty Acids in The
brain sciences Review Importance of the Role of !-3 and !-6 Polyunsaturated Fatty Acids in the Progression of Brain Cancer Mayra Montecillo-Aguado 1,2, Belen Tirado-Rodriguez 2 , Zhen Tong 3,4 , Owen M. Vega 4, Mario Morales-Martínez 2, Shaheen Abkenari 4, José Pedraza-Chaverri 5 and Sara Huerta-Yepez 2,4,* 1 Programa de Doctorado en Ciencias Biomédicas, Facultad de Medicina, Universidad Nacional Autónoma de Mexico (UNAM), Mexico City 04510, Mexico; [email protected] 2 Hospital Infantil de Mexico, Federico Gomez, Unidad de Investigacion en Enfermedades Oncologicas, Mexico City 06720, Mexico; [email protected] (B.T.-R.); [email protected] (M.M.-M.) 3 Molecular Toxicology Interdepartmental Program and Environmental Health Sciences, University of California, Los Angeles, CA 90095, USA; [email protected] 4 Department of Pathology & Laboratory Medicine, University of California, Los Angeles, CA 90095, USA; [email protected] (O.M.V.); [email protected] (S.A.) 5 Departamento de Biología, Facultad de Química, Universidad Nacional Autonoma de Mexico (UNAM), Mexico City 04510, Mexico; [email protected] * Correspondence: [email protected]; Tel.: +52-(55)-52289917 (ext. 4401); Fax: +52-(55)-44349663 Received: 7 May 2020; Accepted: 9 June 2020; Published: 17 June 2020 Abstract: Brain cancer is one of the most malignant types of cancer in both children and adults. Brain cancer patients tend to have a poor prognosis and a high rate of mortality. Additionally, 20–40% of all other types of cancer can develop brain metastasis. Numerous pieces of evidence suggest that omega-3-polyunsaturated fatty acids (!-PUFAs) could potentially be used in the prevention and therapy of several types of cancer. -

Oxylipin Response to Acute and Chronic Exercise: a Systematic Review
H OH metabolites OH Review Oxylipin Response to Acute and Chronic Exercise: A Systematic Review Étore F. Signini 1,* , David C. Nieman 2 , Claudio D. Silva 1 , Camila A. Sakaguchi 1 and Aparecida M. Catai 1 1 Physical Therapy Department, Federal University of São Carlos, São Carlos, SP 13565-905, Brazil; [email protected] (C.D.S.); [email protected] (C.A.S.); [email protected] (A.M.C.) 2 North Carolina Research Campus, Appalachian State University, Kannapolis, NC 28081, USA; [email protected] * Correspondence: [email protected] Received: 28 May 2020; Accepted: 19 June 2020; Published: 25 June 2020 Abstract: Oxylipins are oxidized compounds of polyunsaturated fatty acids that play important roles in the body. Recently, metabololipidomic-based studies using advanced mass spectrometry have measured the oxylipins generated during acute and chronic physical exercise and described the related physiological effects. The objective of this systematic review was to provide a panel of the primary exercise-related oxylipins and their respective functions in healthy individuals. Searches were performed in five databases (Cochrane, PubMed, Science Direct, Scopus and Web of Science) using combinations of the Medical Subject Headings (MeSH) terms: “Humans”, “Exercise”, “Physical Activity”, “Sports”, “Oxylipins”, and “Lipid Mediators”. An adapted scoring system created in a previous study from our group was used to rate the quality of the studies. Nine studies were included after examining 1749 documents. Seven studies focused on the acute effect of physical exercise while two studies determined the effects of exercise training on the oxylipin profile. Numerous oxylipins are mobilized during intensive and prolonged exercise, with most related to the inflammatory process, immune function, tissue repair, cardiovascular and renal functions, and oxidative stress. -

Development and Characterization of Essential Fatty Acid Deficiency In
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 1147-1151, February 1995 Cell Biology Development and characterization of essential fatty acid deficiency in human endothelial cells in culture (eicosapentaenoic acid/prostacycline) RICHARD LERNER*t, PETER LINDSTR6M*, ANDERS BERGt, ELSBETH JOHANSSONt, KERSTIN ROSENDAHLt, AND JAN PALMBLAD* Departments of *Medicine and tClinical Chemistry, Karolinska Institute, Stockholm Soder Hospital, S-118 83 Stockholm, Sweden Communicated by Ralph T. Holman, University of Minnesota, Austin, MN, September 19, 1994 (received for review September 8, 1993) ABSTRACT We induced an essential fatty acid deficiency Against this background we asked whether functional re- (EFAD) in human umbilical vein endothelial cells by culture sponses of endothelial cells, being intimately involved in the in medium with 20% (vol/vol) delipidated fetal calf serum. inflammatory reaction, might be reduced in EFAD. This EFAD, reflected by decreased cellular linoleic acid (18:2co6) report concerns the biochemical characterization of EFAD and arachidonic acid (20:4o6) and emergence ofthe oleic acid produced in human umbilical vein endothelial cells (HUVEC) derivative 5,8,11-eicosatrienoic acid (20:3to9; Mead's acid), by culturing the cells in a delipidated medium, the generation was evident after 1 week of culture and became pronounced of prostacycline (PGI2), and the ability to respond with a rise after 2 weeks. Beyond that time point, control cells (cultured of cytosolic concentrations of Ca2 , [Ca2+] . in 20% normal fetal calf serum) grew deficient of 18:2w6, and EFAD cells died. 18:2co6 addition to EFAD cells resulted in MATERIALS AND METHODS dose-dependent increases of 18:2o6 and 20:4o6. 20:4w6 or 5,8,11,14,17-eicosapentaenoic acid (20:5W3) additions re- Chemicals. -

Aging Alters Lipid Mediator Biosynthesis from Polyunsaturated Fatty Acids in the Brain of Senescence-Accelerated Mice with Age-Related Cognitive Dysfunction
June 2020 Medical Mass Spectrometry Vol. 4 No. 1 Research Paper Aging alters lipid mediator biosynthesis from polyunsaturated fatty acids in the brain of senescence-accelerated mice with age-related cognitive dysfunction Hideyuki Sasaki1,3*, Yoshihisa Kaneda1, Tomohiro Rogi1, Hiroshi Shibata1, Ryohei Aoyagi2, Tsuyoshi Yamane3, Kazutaka Ikeda3, Makoto Arita2,3 1 Institute for Health Care Science, Suntory Wellness Ltd., 8‒1‒1 Seikadai, Seika-cho, Soraku-gun, Kyoto 619‒0284, Japan 2 Division of Physiological Chemistry and Metabolism, Graduate School of Pharmaceutical Sciences, Keio University, 1‒5‒30 Shibakoen, Minato-ku, Tokyo 105‒8512, Japan 3 Laboratory for Metabolomics, RIKEN Center for Integrative Medical Sciences, 1‒7‒22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230‒0045, Japan Abstract This study aimed to clarify whether long-chain polyunsaturated fatty acid (LCPUFA) metabolism in the brain that causes an age-related decline of cognitive function changes with aging. The senescence-accelerated mice-prone 10 (SAMP10) is an animal model used in studies related to aging. Young (two-month-old) mice and aged (nine-month-old) mice were fed control diet for 12 weeks. Age-related changes in lipid mediator profiles in the cortex and hippocampus were assessed by comprehensive lipidomics, using liquid chromatography-tandem mass spectrometry after memory examination. The expression of fatty acid metabolism-related mRNAs was evaluated in the hippocampus. Free arachidonic acid (ARA) levels were extremely high among all detected free fatty acids, being approximately 8-fold higher than those of free DHA in the cortex and hippocampus of both young and aged mice. Level of ARA-derived metabolites was extraordinarily high compared to that of other PUFA-derived metabolites, whereas that of DHA-derived metabolites was very low in the cortex and hippocampus of both mice.