Biamino, Elisa; Di Gregorio, Eleonora
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Transition from Wild to Domesticated Cotton (G
bioRxiv preprint doi: https://doi.org/10.1101/616763; this version posted April 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genetic analysis of the transition from wild to domesticated cotton (G. hirsutum) Corrinne E. Grover1, Mi-Jeong Yoo1,a, Michael A. Gore2, David B. Harker3,b, Robert L. Byers3, Alexander E. Lipka4, Guanjing Hu1, Daojun Yuan1, Justin L. Conover1, Joshua A. Udall3,c, Andrew H. Paterson5, and Jonathan F. Wendel1 1Department of Ecology, Evolution and Organismal Biology, Iowa State University, Ames, IA 50011 2Plant Breeding and Genetics Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853 3Plant and Wildlife Science Department, Brigham Young University, Provo, UT 84602 4Department of Crop Sciences, University of Illinois, Urbana, IL 61801 5Plant Genome Mapping Laboratory, University of Georgia, Athens, GA 30602 acurrent address: Department of Biology, University of Florida, Gainesville, FL 32611 bcurrent address: Department of Dermatology, The University of Texas Southwestern Medical Center, Dallas, TX 75390 ccurrent address: Southern Plains Agricultural Research Center, USDA-ARS, College Station, TX, 77845- 4988 Corresponding Author: Jonathan F. Wendel ([email protected]) Data Availability: All data are available via figshare (https://figshare.com/projects/Genetic_analysis_of_the_transition_from_wild_to_domesticated_cotton_G _hirsutum_QTL/62693) and GitHub (https://github.com/Wendellab/QTL_TxMx). Keywords: QTL, domestication, Gossypium hirsutum, cotton Summary: An F2 population between truly wild and domesticated cotton was used to identify QTL associated with selection under domestication. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Clinical Application of Chromosomal Microarray Analysis for Fetuses With
Xu et al. Molecular Cytogenetics (2020) 13:38 https://doi.org/10.1186/s13039-020-00502-5 RESEARCH Open Access Clinical application of chromosomal microarray analysis for fetuses with craniofacial malformations Chenyang Xu1†, Yanbao Xiang1†, Xueqin Xu1, Lili Zhou1, Huanzheng Li1, Xueqin Dong1 and Shaohua Tang1,2* Abstract Background: The potential correlations between chromosomal abnormalities and craniofacial malformations (CFMs) remain a challenge in prenatal diagnosis. This study aimed to evaluate 118 fetuses with CFMs by applying chromosomal microarray analysis (CMA) and G-banded chromosome analysis. Results: Of the 118 cases in this study, 39.8% were isolated CFMs (47/118) whereas 60.2% were non-isolated CFMs (71/118). The detection rate of chromosomal abnormalities in non-isolated CFM fetuses was significantly higher than that in isolated CFM fetuses (26/71 vs. 7/47, p = 0.01). Compared to the 16 fetuses (16/104; 15.4%) with pathogenic chromosomal abnormalities detected by karyotype analysis, CMA identified a total of 33 fetuses (33/118; 28.0%) with clinically significant findings. These 33 fetuses included cases with aneuploidy abnormalities (14/118; 11.9%), microdeletion/microduplication syndromes (9/118; 7.6%), and other pathogenic copy number variations (CNVs) only (10/118; 8.5%).We further explored the CNV/phenotype correlation and found a series of clear or suspected dosage-sensitive CFM genes including TBX1, MAPK1, PCYT1A, DLG1, LHX1, SHH, SF3B4, FOXC1, ZIC2, CREBBP, SNRPB, and CSNK2A1. Conclusion: These findings enrich our understanding of the potential causative CNVs and genes in CFMs. Identification of the genetic basis of CFMs contributes to our understanding of their pathogenesis and allows detailed genetic counselling. -

Genetic Testing: Background and Policy Issues
Genetic Testing: Background and Policy Issues Amanda K. Sarata Specialist in Health Policy March 2, 2015 Congressional Research Service 7-5700 www.crs.gov RL33832 Genetic Testing: Background and Policy Issues Summary Congress has considered, at various points in time, numerous pieces of legislation that relate to genetic and genomic technology and testing. These include bills addressing genetic discrimination in health insurance and employment; precision medicine; the patenting of genetic material; and the oversight of clinical laboratory tests (in vitro diagnostics), including genetic tests. The focus on these issues signals the growing importance of public policy issues surrounding the clinical and public health implications of new genetic technology. As genetic technologies proliferate and are increasingly used to guide clinical treatment, these public policy issues are likely to continue to garner attention. Understanding the basic scientific concepts underlying genetics and genetic testing may help facilitate the development of more effective public policy in this area. Humans have 23 pairs of chromosomes in the nucleus of most cells in their bodies. Chromosomes are composed of deoxyribonucleic acid (DNA) and protein. DNA is composed of complex chemical substances called bases. Proteins are fundamental components of all living cells, and include enzymes, structural elements, and hormones. A gene is the section of DNA that contains the sequence which corresponds to a specific protein. Though most of the genome is similar between individuals, there can be significant variation in physical appearance or function between individuals due to variations in DNA sequence that may manifest as changes in the protein, which affect the protein’s function. -

Identification of a Novel Deletion Region in 3Q29 Microdeletion Syndrome by Oligonucleotide Array Comparative Genomic Hybridization
Korean J Lab Med 2010;30:70-5 � Original Article∙Diagnostic Genetics � DOI 10.3343/kjlm.2010.30.1.70 Identification of a Novel Deletion Region in 3q29 Microdeletion Syndrome by Oligonucleotide Array Comparative Genomic Hybridization Eul-Ju Seo, M.D.1, Kyung Ran Jun, M.D.1, Han-Wook Yoo, M.D.2, Hanik K. Yoo, M.D.3, and Jin-Ok Lee, M.S.4 Departments of Laboratory Medicine1, Pediatrics2, and Psychiatry3, University of Ulsan College of Medicine and Asan Medical Center, Seoul; Asan Institute for Life Sciences4, Seoul, Korea Background : The 3q29 microdeletion syndrome is a genomic disorder characterized by mental retardation, developmental delay, microcephaly, and slight facial dysmorphism. In most cases, the microdeletion spans a 1.6-Mb region between low-copy repeats (LCRs). We identified a novel 4.0- Mb deletion using oligonucleotide array comparative genomic hybridization (array CGH) in monozy- gotic twin sisters. Methods : G-banded chromosome analysis was performed in the twins and their parents. High- resolution oligonucleotide array CGH was performed using the human whole genome 244K CGH microarray (Agilent Technologies, USA) followed by validation using FISH, and the obtained results were analyzed using the genome database resources. Results : G-banding revealed that the twins had de novo 46,XX,del(3)(q29) karyotype. Array CGH showed a 4.0-Mb interstitial deletion on 3q29, which contained 39 genes and no breakpoints flanked by LCRs. In addition to the typical characteristics of the 3q29 microdeletion syndrome, the twins had attention deficit-hyperactivity disorder, strabismus, congenital heart defect, and gray hair. Besides the p21-activated protein kinase (PAK2) and discs large homolog 1 (DLG1) genes, which are known to play a critical role in mental retardation, the hairy and enhancer of split 1 (HES1) and antigen p97 (melanoma associated; MFI2) genes might be possible candidate genes associated with strabis- mus, congenital heart defect, and gray hair. -

Table SII. Significantly Differentially Expressed Mrnas of GSE23558 Data Series with the Criteria of Adjusted P<0.05 And
Table SII. Significantly differentially expressed mRNAs of GSE23558 data series with the criteria of adjusted P<0.05 and logFC>1.5. Probe ID Adjusted P-value logFC Gene symbol Gene title A_23_P157793 1.52x10-5 6.91 CA9 carbonic anhydrase 9 A_23_P161698 1.14x10-4 5.86 MMP3 matrix metallopeptidase 3 A_23_P25150 1.49x10-9 5.67 HOXC9 homeobox C9 A_23_P13094 3.26x10-4 5.56 MMP10 matrix metallopeptidase 10 A_23_P48570 2.36x10-5 5.48 DHRS2 dehydrogenase A_23_P125278 3.03x10-3 5.40 CXCL11 C-X-C motif chemokine ligand 11 A_23_P321501 1.63x10-5 5.38 DHRS2 dehydrogenase A_23_P431388 2.27x10-6 5.33 SPOCD1 SPOC domain containing 1 A_24_P20607 5.13x10-4 5.32 CXCL11 C-X-C motif chemokine ligand 11 A_24_P11061 3.70x10-3 5.30 CSAG1 chondrosarcoma associated gene 1 A_23_P87700 1.03x10-4 5.25 MFAP5 microfibrillar associated protein 5 A_23_P150979 1.81x10-2 5.25 MUCL1 mucin like 1 A_23_P1691 2.71x10-8 5.12 MMP1 matrix metallopeptidase 1 A_23_P350005 2.53x10-4 5.12 TRIML2 tripartite motif family like 2 A_24_P303091 1.23x10-3 4.99 CXCL10 C-X-C motif chemokine ligand 10 A_24_P923612 1.60x10-5 4.95 PTHLH parathyroid hormone like hormone A_23_P7313 6.03x10-5 4.94 SPP1 secreted phosphoprotein 1 A_23_P122924 2.45x10-8 4.93 INHBA inhibin A subunit A_32_P155460 6.56x10-3 4.91 PICSAR P38 inhibited cutaneous squamous cell carcinoma associated lincRNA A_24_P686965 8.75x10-7 4.82 SH2D5 SH2 domain containing 5 A_23_P105475 7.74x10-3 4.70 SLCO1B3 solute carrier organic anion transporter family member 1B3 A_24_P85099 4.82x10-5 4.67 HMGA2 high mobility group AT-hook 2 A_24_P101651 -

Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency
Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. A Maddalena, … , W E O'Brien, R L Nussbaum J Clin Invest. 1988;82(4):1353-1358. https://doi.org/10.1172/JCI113738. Research Article Point mutations in the X-linked ornithine transcarbamylase (OTC) gene have been detected at the same Taq I restriction site in 3 of 24 unrelated probands with OTC deficiency. A de novo mutation could be traced in all three families to an individual in a prior generation, confirming independent recurrence. The DNA sequence in the region of the altered Taq I site was determined in the three probands. In two unrelated male probands with neonatal onset of severe OTC deficiency, a guanine (G) to adenine (A) mutation on the sense strand (antisense cytosine [C] to thymine [T]) was found, resulting in glutamine for arginine at amino acid 109 of the mature polypeptide. In the third case, where the proband was a symptomatic female, C to T (sense strand) transition converted residue 109 to a premature stop. These results support the observation that Taq I restriction sites, which contain an internal CG, are particularly susceptible to C to T transition mutation due to deamination of a methylated C in either the sense or antisense strand. The OTC gene seems especially sensitive to C to T transition mutation at arginine codon 109 because either a nonsense mutation or an extremely deleterious missense mutation will result. Find the latest version: https://jci.me/113738/pdf Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency Anne Maddalena,*" J. -

Indel Information Eliminates Trivial Sequence Alignment in Maximum Likelihood Phylogenetic Analysis
Cladistics Cladistics 28 (2012) 514–528 10.1111/j.1096-0031.2012.00402.x Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis John S.S. Dentona,b,* and Ward C. Wheelerb,c aDivision of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA; bRichard Gilder Graduate School, American Museum of Natural History, New York, NY 10024, USA; cDivision of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA Accepted 12 March 2012 Abstract Although there has been a recent proliferation in maximum-likelihood (ML)-based tree estimation methods based on a fixed sequence alignment (MSA), little research has been done on incorporating indel information in this traditional framework. We show, using a simple model on a single character example, that a trivial alignment of a different form than that previously identified for parsimony is optimal in ML under standard assumptions treating indels as ‘‘missing’’ data, but that it is not optimal when indels are incorporated into the character alphabet. We show that the optimality of the trivial alignment is not an artefact of simplified theory assumptions by demonstrating that trivial alignment likelihoods of five different multiple sequence alignment datasets exhibit this phenomenon. These results demonstrate the need for use of indel information in likelihood analysis on fixed MSAs, and suggest that caution must be exercised when drawing conclusions from software implementations claiming improvements in likelihood scores under an indels-as-missing assumption. Ó The Willi Hennig Society 2012. Maximum likelihood (ML) has become a popular the increasing sophistication of search heuristics in ML optimality criterion for inferring evolutionary trees software (e.g. -
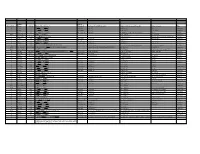
Systematic (Genomic Based)
Trivial Codon Exon/Intron Name Codon Nucleotide Change* Changed Systematic(cDNA based) Systematic (genomic based) Other names Type 2 M-1T -1 CCATGGC>CCACGGC Met>Thr c.2T>C g.4895T>C M1T transition 2 R3Op 3 CACCGAT>CACTGAT Arg>Opal c.10C>T g.4903C>T R3X, R3ter transition 2 ∆A20 20 CCC[A]GAG>CCCGAG Gln>Arg (fs) c.62delA g.4955delA (first base of deletion) ∆A20 deletion IVS2 IVS2-1G>A atagGTA>ataaGTA g.5814G>A c.113-1G>A transition 3 IVS2-1∆4 37-38 ata[gGTA]CCA>ataCCA g.5813delGGTA G12/∆3;IVS2-delGGTA;∆4IVS2-E3 deletion 3 R59op 59 TTCCGAG>TTCTGAG Arg>Opal c.178C>T g.5880C>T R59X;R59ter transition 3 I73T 73 GCATCGG>GCACCGG Ile>Thr c.221T>C g.5923T>C transition 3 L83∆C 83 CCT[C]TAC>CCTTAC Leu>Leu(fs) c.252delC g.5954delC c.250 delC deletion 3 1123ins12 104 GGT[]GGG>GGT[GGGGATCGTGGT]GGG c.314^315ins12 g.6016^6017insGGGGATCGTGGT —12E3;V104+GIVV insertion IVS3 K107K∆12 104-107del GTG[GTGGGAATCAAG]gtt>GTGgtgggaatcaaagtt c.324G>A;delGTGGGAATCAAG(312-324) g.6026G>A g.1133G>A transition IVS3 IVS3-1G>A acagTTA>acaaTTA g.7257G>A c.325-1G>C, g.8180G>C,IVS3sas transversion 4 ∆28E4 114-151 TCC[TCTTGCAGGAACAAACAAAGAAACCACC]ATT>TCCATT Pro>Pro(fs) c.345-372del28 g.7277^7305del c.345_72del28 deletion 4 Q110Oc 110 GACCAAG>GACTAAG Gln>Ochre c.331C>T g.7264C>T transition 4 ∆4E4 118-120 GAAC[AAAC]AAAG>GAACAAAG c.357delAAAC g.7289delAAAC ∆4, MD∆4 deletion 4-5 ∆E4-E5 ccc[tgt…TCC]AGC>cccAGC g.6594^8239del F13/∆4,5 deletion 5 C134R 134 CGCTGTG>CGCCGTG Cys>Arg c.403T>C g.8162T>C C135R transition 5 W147R 147 AAGTGGC>AAGCGGC Trp>Arg c.442T>C g.8201T>C W148R transition -

A Molecular Quantitative Trait Locus Map for Osteoarthritis
A molecular quantitative trait locus map for osteoarthritis Julia Steinberg1,2,3,4, Lorraine Southam1,3, Theodoros I Roumeliotis3,5, Matthew J Clark6, Raveen L Jayasuriya6, Diane Swift6, Karan M Shah6, Natalie C Butterfield7, Roger A Brooks8, Andrew W McCaskie8, JH Duncan Bassett7, Graham R Williams7, Jyoti S Choudhary3,5, J Mark Wilkinson6,9,11, Eleftheria Zeggini1,3,10,11 1 Institute of Translational Genomics, Helmholtz Zentrum München – German Research Center for Environmental Health, 85764 Neuherberg, Germany 2 Cancer Research Division, Cancer Council NSW, Sydney, New South Wales 2011, Australia 3 Wellcome Sanger Institute, Hinxton CB10 1SA, United Kingdom 4 School of Public Health, The University of Sydney, Sydney, New South Wales 2006, Australia 5 The Institute of Cancer Research, London SW7 3RP, UK 6 Department of Oncology and Metabolism, University of Sheffield, Sheffield S10 2RX, UK 7 Molecular Endocrinology Laboratory, Department of Metabolism, Digestion and Reproduction, Imperial College London, London W12 ONN, UK 8 Division of Trauma & Orthopaedic Surgery, Department of Surgery, University of Cambridge, Cambridge CB2 2QQ, UK 9 Centre for Integrated Research into Musculoskeletal Ageing and Sheffield Healthy Lifespan Institute, University of Sheffield, Sheffield S10 2TN, UK 10 TUM School of Medicine, Technical University of Munich and Klinikum Rechts der Isar, Munich, Germany 11 These authors contributed equally Correspondence to: [email protected]; eleftheria.zeggini@helmholtz- muenchen.de Supplementary Information Table of Contents Supplementary Notes ................................................................................................................... 3 Supplementary Note 1: Identification of cis-eQTLs and cis-pQTLs in osteoarthritis tissues ..................... 3 Supplementary Note 2: Differential eQTLs between low-grade and high-grade cartilage ....................... 4 Supplementary Note 3: Differential gene expression between high-grade and low-grade cartilage ...... -

G14TBS Part II: Population Genetics
G14TBS Part II: Population Genetics Dr Richard Wilkinson Room C26, Mathematical Sciences Building Please email corrections and suggestions to [email protected] Spring 2015 2 Preliminaries These notes form part of the lecture notes for the mod- ule G14TBS. This section is on Population Genetics, and contains 14 lectures worth of material. During this section will be doing some problem-based learning, where the em- phasis is on you to work with your classmates to generate your own notes. I will be on hand at all times to answer questions and guide the discussions. Reading List: There are several good introductory books on population genetics, although I found their level of math- ematical sophistication either too low or too high for the purposes of this module. The following are the sources I used to put together these lecture notes: • Gillespie, J. H., Population genetics: a concise guide. John Hopkins University Press, Baltimore and Lon- don, 2004. • Hartl, D. L., A primer of population genetics, 2nd edition. Sinauer Associates, Inc., Publishers, 1988. • Ewens, W. J., Mathematical population genetics: (I) Theoretical Introduction. Springer, 2000. • Holsinger, K. E., Lecture notes in population genet- ics. University of Connecticut, 2001-2010. Available online at http://darwin.eeb.uconn.edu/eeb348/lecturenotes/notes.html. • Tavar´eS., Ancestral inference in population genetics. In: Lectures on Probability Theory and Statistics. Ecole d'Et´esde Probabilit´ede Saint-Flour XXXI { 2001. (Ed. Picard J.) Lecture Notes in Mathematics, Springer Verlag, 1837, 1-188, 2004. Available online at http://www.cmb.usc.edu/people/stavare/STpapers-pdf/T04.pdf Gillespie, Hartl and Holsinger are written for non-mathematicians and are easiest to follow.