Spatially Clustered Loci with Multiple Enhancers Are Frequent Targets of HIV-1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Dominance) Effects of Genetic Variants Associated with Refractive Error And
Molecular Genetics and Genomics https://doi.org/10.1007/s00438-020-01666-w ORIGINAL ARTICLE Non‑additive (dominance) efects of genetic variants associated with refractive error and myopia Alfred Pozarickij1 · Cathy Williams2 · Jeremy A. Guggenheim1 · and the UK Biobank Eye and Vision Consortium Received: 26 November 2019 / Accepted: 16 March 2020 © The Author(s) 2020 Abstract Genome-wide association studies (GWAS) have revealed that the genetic contribution to certain complex diseases is well- described by Fisher’s infnitesimal model in which a vast number of polymorphisms each confer a small efect. Under Fisher’s model, variants have additive efects both across loci and within loci. However, the latter assumption is at odds with the com- mon observation of dominant or recessive rare alleles responsible for monogenic disorders. Here, we searched for evidence of non-additive (dominant or recessive) efects for GWAS variants known to confer susceptibility to the highly heritable quantitative trait, refractive error. Of 146 GWAS variants examined in a discovery sample of 228,423 individuals whose refractive error phenotype was inferred from their age-of-onset of spectacle wear, only 8 had even nominal evidence (p < 0.05) of non-additive efects. In a replication sample of 73,577 individuals who underwent direct assessment of refractive error, 1 of these 8 variants had robust independent evidence of non-additive efects (rs7829127 within ZMAT4, p = 4.76E−05) while a further 2 had suggestive evidence (rs35337422 in RD3L, p = 7.21E−03 and rs12193446 in LAMA2, p = 2.57E−02). Account- ing for non-additive efects had minimal impact on the accuracy of a polygenic risk score for refractive error (R2 = 6.04% vs. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Structural Insights Into Ubiquitin Recognition and Ufd1 Interaction of Npl4
ARTICLE https://doi.org/10.1038/s41467-019-13697-y OPEN Structural insights into ubiquitin recognition and Ufd1 interaction of Npl4 Yusuke Sato1,2,3,5,7, Hikaru Tsuchiya4,7, Atsushi Yamagata1,2,3,6, Kei Okatsu1,2, Keiji Tanaka4, Yasushi Saeki 4* & Shuya Fukai 1,2,3* Npl4 is likely to be the most upstream factor recognizing Lys48-linked polyubiquitylated substrates in the proteasomal degradation pathway in yeast. Along with Ufd1, Npl4 forms a 1234567890():,; heterodimer (UN), and functions as a cofactor for the Cdc48 ATPase. Here, we report the crystal structures of yeast Npl4 in complex with Lys48-linked diubiquitin and with the Npl4- binding motif of Ufd1. The distal and proximal ubiquitin moieties of Lys48-linked diubiquitin primarily interact with the C-terminal helix and N-terminal loop of the Npl4 C-terminal domain (CTD), respectively. Mutational analysis suggests that the CTD contributes to linkage selectivity and initial binding of ubiquitin chains. Ufd1 occupies a hydrophobic groove of the Mpr1/Pad1 N-terminal (MPN) domain of Npl4, which corresponds to the catalytic groove of the MPN domain of JAB1/MPN/Mov34 metalloenzyme (JAMM)-family deubiquitylating enzyme. This study provides important structural insights into the polyubiquitin chain recognition by the Cdc48–UN complex and its assembly. 1 Institute for Quantitative Biosciences, The University of Tokyo, Tokyo 113-0032, Japan. 2 Synchrotron Radiation Research Organization, The University of Tokyo, Tokyo 113-0032, Japan. 3 Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Chiba 277-8562, Japan. 4 Laboratory of Protein Metabolism, Tokyo Metropolitan Institute of Medical Science, Tokyo 156-8506, Japan. -

WIPI-1 (38-W): Sc-100901
SANTA CRUZ BIOTECHNOLOGY, INC. WIPI-1 (38-W): sc-100901 BACKGROUND APPLICATIONS WIPI-1 (WD repeat domain, phosphoinositide interacting-1), also known as WIPI-1 (38-W) is recommended for detection of WIPI-1 of mouse, rat and WIPI1, ATG18 or WIPI49, is a 446 amino acid protein that localizes to cyto- human origin by Western Blotting (starting dilution 1:200, dilution range plasmic vesicles, endosomes, clathrin-coated vesicles and the trans-Golgi 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein network. Ubiquitously expressed with highest expression in heart, testis, (1 ml of cell lysate)] and solid phase ELISA (starting dilution 1:30, dilution placenta, pancreas and skeletal muscle, WIPI-1 is thought to play a role in range 1:30-1:3000). autophagy and may regulate protein trafficking in certain recycling pathways. Suitable for use as control antibody for WIPI-1 siRNA (h): sc-72210, WIPI-1 In addition, WIPI-1 interacts with androgen and estrogen receptors (ARs and siRNA (m): sc-72211, WIPI-1 shRNA Plasmid (h): sc-72210-SH, WIPI-1 shRNA ERs, respectively) and, through this interaction, may modify receptor function. Plasmid (m): sc-72211-SH, WIPI-1 shRNA (h) Lentiviral Particles: sc-72210-V WIPI-1 contains three WD repeats and has a 7-bladed propeller structure and WIPI-1 shRNA (m) Lentiviral Particles: sc-72211-V. with a conserved motif that facilitates its interaction with other proteins. WIPI-1 is expressed as two isoforms, designated a and b, and its expression Molecular Weight of WIPI-1: 49 kDa. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Allele-Specific Alternative Splicing and Its Functional Genetic Variants in Human Tissues
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Allele-specific alternative splicing and its functional genetic variants in human tissues Kofi Amoah1,†, Yun-Hua Esther Hsiao2,†, Jae Hoon Bahn3†, Yiwei Sun3, Christina Burghard1, Boon Xin Tan3, Ei-Wen Yang3, Xinshu Xiao1,2,3,4,5,* 1Bioinformatics Interdepartmental Program, 2Department of Bioengineering, 3Department of Integrative Biology and Physiology, 4Molecular Biology Institute, 5Institute for Quantitative and Computational Biosciences, University of California, Los Angeles, CA 90095, USA. † These authors contributed equally. *Correspondence: [email protected] 1 Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Alternative splicing is an RNA processing mechanism that affects most genes in human, contributing to disease mechanisms and phenotypic diversity. The regulation of splicing involves an intricate network of cis-regulatory elements and trans-acting factors. Due to their high sequence specificity, cis-regulation of splicing can be altered by genetic variants, significantly affecting splicing outcomes. Recently, multiple methods have been applied to understanding the regulatory effects of genetic variants on splicing. However, it is still challenging to go beyond apparent association to pinpoint functional variants. To fill in this gap, we utilized large-scale datasets of the Genotype-Tissue Expression (GTEx) project to study genetically-modulated alternative splicing (GMAS) via identification of allele-specific splicing events. We demonstrate that GMAS events are shared across tissues and individuals more often than expected by chance, consistent with their genetically driven nature. Moreover, although the allelic bias of GMAS exons varies across samples, the degree of variation is similar across tissues vs. -

Genome-Wide Association Study of Cardiac Structure and Systolic Function in African Americans the Candidate Gene Association Resource (Care) Study Ervin R
Genome-Wide Association Study of Cardiac Structure and Systolic Function in African Americans The Candidate Gene Association Resource (CARe) Study Ervin R. Fox, University of Mississippi Solomon K. Musani, University of Mississippi Maja Barbalic, University of Texas Health Science Center Honghuang Lin, Boston University Bing Yu, University of Mississippi Kofo O. Ogunyankin, Northwestern University Nicholas L. Smith, University of Washington Abdullah Kutlar, Georgia Health Sciences University Nicole L. Glazer, Boston University Wendy S. Post, Johns Hopkins University Only first 10 authors above; see publication for full author list. Journal Title: Circulation: Cardiovascular Genetics Volume: Volume 6, Number 1 Publisher: American Heart Association | 2013-02-01, Pages 37-46 Type of Work: Article | Post-print: After Peer Review Publisher DOI: 10.1161/CIRCGENETICS.111.962365 Permanent URL: https://pid.emory.edu/ark:/25593/v8g7t Final published version: http://dx.doi.org/10.1161/CIRCGENETICS.111.962365 Copyright information: © 2013 American Heart Association, Inc. Accessed September 30, 2021 9:00 PM EDT NIH Public Access Author Manuscript Circ Cardiovasc Genet. Author manuscript; available in PMC 2014 February 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Circ Cardiovasc Genet. 2013 February 1; 6(1): 37–46. doi:10.1161/CIRCGENETICS.111.962365. Genome-Wide Association Study of Cardiac Structure and Systolic Function in African Americans: The Candidate Gene Association Resource (CARe) Study Ervin R. Fox, MD1,*, Solomon K. Musani, PhD1,*, Maja Barbalic, PhD2,*, Honghuang Lin, PhD3, Bing Yu, MS1, Kofo O. Ogunyankin, MD4, Nicholas L. Smith, PhD5, Abdullah Kutlar, MD6, Nicole L. Glazer, MD3, Wendy S. -
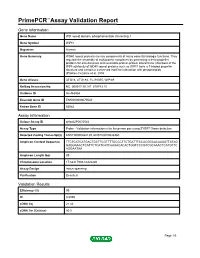
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name WD repeat domain, phosphoinositide interacting 1 Gene Symbol WIPI1 Organism Human Gene Summary WD40 repeat proteins are key components of many essential biologic functions. They regulate the assembly of multiprotein complexes by presenting a beta-propeller platform for simultaneous and reversible protein-protein interactions. Members of the WIPI subfamily of WD40 repeat proteins such as WIPI1 have a 7-bladed propeller structure and contain a conserved motif for interaction with phospholipids (Proikas-Cezanne et al. 2004 Gene Aliases ATG18, ATG18A, FLJ10055, WIPI49 RefSeq Accession No. NC_000017.10, NT_010783.15 UniGene ID Hs.463964 Ensembl Gene ID ENSG00000070540 Entrez Gene ID 55062 Assay Information Unique Assay ID qHsaCIP0030544 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000262139, ENST00000546360 Amplicon Context Sequence TTCTCATCATGACTGCTTCGTTTTGCCCTTCTGATTTCCACGGCACAAGATTATAG GAGGAAACTCATTCTCATCATCAAGACACACTGGTCCCGTCGCAAACTCATGTTC AGGAATAA Amplicon Length (bp) 89 Chromosome Location 17:66417908-66422280 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9995 cDNA Cq 21.43 cDNA Tm (Celsius) 80.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 35.07 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report WIPI1, Human Amplification Plot Amplification of cDNA -

A Multiethnic Genome-Wide Analysis of 44,039 Individuals Identifies 41 New Loci Associated with Central Corneal Thickness
ARTICLE https://doi.org/10.1038/s42003-020-1037-7 OPEN A multiethnic genome-wide analysis of 44,039 individuals identifies 41 new loci associated with central corneal thickness ✉ Hélène Choquet 1 , Ronald B. Melles2, Jie Yin1, Thomas J. Hoffmann 3,4, Khanh K. Thai1, Mark N. Kvale3, 1234567890():,; Yambazi Banda3, Alison J. Hardcastle 5,6, Stephen J. Tuft7, M. Maria Glymour4, Catherine Schaefer 1, ✉ Neil Risch1,3,4, K. Saidas Nair8, Pirro G. Hysi 9,10,11 & Eric Jorgenson 1 Central corneal thickness (CCT) is one of the most heritable human traits, with broad-sense heritability estimates ranging between 0.68 to 0.95. Despite the high heritability and numerous previous association studies, only 8.5% of CCT variance is currently explained. Here, we report the results of a multiethnic meta-analysis of available genome-wide asso- ciation studies in which we find association between CCT and 98 genomic loci, of which 41 are novel. Among these loci, 20 were significantly associated with keratoconus, and one (RAPSN rs3740685) was significantly associated with glaucoma after Bonferroni correction. Two-sample Mendelian randomization analysis suggests that thinner CCT does not causally increase the risk of primary open-angle glaucoma. This large CCT study explains up to 14.2% of CCT variance and increases substantially our understanding of the etiology of CCT var- iation. This may open new avenues of investigation into human ocular traits and their rela- tionship to the risk of vision disorders. 1 Kaiser Permanente Northern California (KPNC), Division of Research, Oakland, CA 94612, USA. 2 KPNC, Department of Ophthalmology, Redwood City, CA 94063, USA. -

A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants
A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Lars G. Fritsche1†, Wilmar Igl2†, Jessica N. Cooke Bailey3†, Felix Grassmann4†, Sebanti Sengupta1†, Jennifer L. Bragg-Gresham1,5, Kathryn P. Burdon6, Scott J. Hebbring7, Cindy Wen8, Mathias Gorski2, Ivana K. Kim9, David Cho10, Donald Zack11,12,13,14,15, Eric Souied16, Hendrik P. N. Scholl11,17, Elisa Bala18, Kristine E. Lee19, David J. Hunter20,21, Rebecca J. Sardell22, Paul Mitchell23, Joanna E. Merriam24, Valentina Cipriani25,26, Joshua D. Hoffman27, Tina Schick28, Yara T. E. Lechanteur29, Robyn H. Guymer30, Matthew P. Johnson31, Yingda Jiang32, Chloe M. Stanton33, Gabriëlle H. S. Buitendijk34,35, Xiaowei Zhan1,36,37, Alan M. Kwong1, Alexis Boleda38, Matthew Brooks39, Linn Gieser38, Rinki Ratnapriya38, Kari E. Branham39, Johanna R. Foerster1, John R. Heckenlively39, Mohammad I. Othman39, Brendan J. Vote6, Helena Hai Liang30, Emmanuelle Souzeau40, Ian L. McAllister41, Timothy Isaacs41, Janette Hall40, Stewart Lake40, David A. Mackey6,30,41, Ian J. Constable41, Jamie E. Craig40, Terrie E. Kitchner7, Zhenglin Yang42,43, Zhiguang Su44, Hongrong Luo8,44, Daniel Chen8, Hong Ouyang8, Ken Flagg8, Danni Lin8, Guanping Mao8, Henry Ferreyra8, Klaus Stark2, Claudia N. von Strachwitz45, Armin Wolf46, Caroline Brandl2,4,47, Guenther Rudolph46, Matthias Olden2, Margaux A. Morrison48, Denise J. Morgan48, Matthew Schu49,50,51,52,53, Jeeyun Ahn54, Giuliana Silvestri55, Evangelia E. Tsironi56, Kyu Hyung Park57, Lindsay A. Farrer49,50,51,52,53, Anton Orlin58, Alexander Brucker59, Mingyao Li60, Christine Curcio61, Saddek Mohand-Saïd62,63,64,65, José-Alain Sahel62,63,64,65,66,67,68, Isabelle Audo62,63,64,69, Mustapha Benchaboune65, Angela J. -

Targeting Melanoma-Initiating Cells by Caffeine: in Silico and in Vitro Approaches
molecules Article Targeting Melanoma-Initiating Cells by Caffeine: In Silico and In Vitro Approaches Claudio Tabolacci 1,* , Martina Cordella 1, Stefania Rossi 1 , Marialaura Bonaccio 2 , Adriana Eramo 1, Carlo Mischiati 3 , Simone Beninati 4 , Licia Iacoviello 2,5 , Antonio Facchiano 6,† and Francesco Facchiano 1,† 1 Department of Oncology and Molecular Medicine, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy; [email protected] (M.C.); [email protected] (S.R.); [email protected] (A.E.); [email protected] (F.F.) 2 Department of Epidemiology and Prevention, IRCCS Neuromed, 86077 Pozzilli, Italy; [email protected] (M.B.); [email protected] (L.I.) 3 Department of Neuroscience and Rehabilitation, School of Medicine, University of Ferrara, 44121 Ferrara, Italy; [email protected] 4 Department of Biology, University of Rome Tor Vergata, 00133 Rome, Italy; [email protected] 5 Department of Medicine and Surgery, Research Center in Epidemiology and Preventive Medicine (EPIMED), University of Insubria, 21100 Varese-Como, Italy 6 Laboratory of Molecular Oncology, IDI-IRCCS, 00167 Rome, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: The beneficial effects of coffee on human diseases are well documented, but the molecular mechanisms of its bioactive compounds on cancer are not completely elucidated. This is likely due to the large heterogeneity of coffee preparations and different coffee-based beverages, but also to Citation: Tabolacci, C.; Cordella, M.; the choice of experimental models where proliferation, differentiation and immune responses are Rossi, S.; Bonaccio, M.; Eramo, A.; differently affected. -

A Commonly Occurring Genetic Variant Within the NPLOC4–TSPAN10–PDE6G Gene Cluster Is Associated with the Risk of Strabismus
Human Genetics https://doi.org/10.1007/s00439-019-02022-8 ORIGINAL INVESTIGATION A commonly occurring genetic variant within the NPLOC4–TSPAN10–PDE6G gene cluster is associated with the risk of strabismus Denis Plotnikov1 · Rupal L. Shah1 · Jamille N. Rodrigues2 · Phillippa M. Cumberland3,4 · Jugnoo S. Rahi3,4,5,6 · Pirro G. Hysi7 · Denize Atan2 · Cathy Williams2 · Jeremy A. Guggenheim1 · UK Biobank Eye and Vision Consortium Received: 20 December 2018 / Accepted: 20 April 2019 © The Author(s) 2019 Abstract Strabismus refers to an abnormal alignment of the eyes leading to the loss of central binocular vision. Concomitant strabis- mus occurs when the angle of deviation is constant in all positions of gaze and often manifests in early childhood when it is considered to be a neurodevelopmental disorder of the visual system. As such, it is inherited as a complex genetic trait, afecting 2–4% of the population. A genome-wide association study (GWAS) for self-reported strabismus (1345 cases and 65,349 controls from UK Biobank) revealed a single genome-wide signifcant locus on chromosome 17q25. Approximately 20 variants across the NPLOC4–TSPAN10–PDE6G gene cluster and in almost perfect linkage disequilibrium (LD) were most strongly associated (lead variant: rs75078292, OR = 1.26, p = 2.24E−08). A recessive model provided a better ft to the data than an additive model. Association with strabismus was independent of refractive error, and the degree of association with strabismus was minimally attenuated after adjustment for amblyopia. The association with strabismus was replicated in an independent cohort of clinician-diagnosed children aged 7 years old (116 cases and 5084 controls; OR = 1.85, p = 0.009).