1 an Introduction to Chirality at the Nanoscale Laurence D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chirality-Assisted Lateral Momentum Transfer for Bidirectional
Shi et al. Light: Science & Applications (2020) 9:62 Official journal of the CIOMP 2047-7538 https://doi.org/10.1038/s41377-020-0293-0 www.nature.com/lsa ARTICLE Open Access Chirality-assisted lateral momentum transfer for bidirectional enantioselective separation Yuzhi Shi 1,2, Tongtong Zhu3,4,5, Tianhang Zhang3, Alfredo Mazzulla 6, Din Ping Tsai 7,WeiqiangDing 5, Ai Qun Liu 2, Gabriella Cipparrone6,8, Juan José Sáenz9 and Cheng-Wei Qiu 3 Abstract Lateral optical forces induced by linearly polarized laser beams have been predicted to deflect dipolar particles with opposite chiralities toward opposite transversal directions. These “chirality-dependent” forces can offer new possibilities for passive all-optical enantioselective sorting of chiral particles, which is essential to the nanoscience and drug industries. However, previous chiral sorting experiments focused on large particles with diameters in the geometrical-optics regime. Here, we demonstrate, for the first time, the robust sorting of Mie (size ~ wavelength) chiral particles with different handedness at an air–water interface using optical lateral forces induced by a single linearly polarized laser beam. The nontrivial physical interactions underlying these chirality-dependent forces distinctly differ from those predicted for dipolar or geometrical-optics particles. The lateral forces emerge from a complex interplay between the light polarization, lateral momentum enhancement, and out-of-plane light refraction at the particle-water interface. The sign of the lateral force could be reversed by changing the particle size, incident angle, and polarization of the obliquely incident light. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction interface15,16. The displacements of particles controlled by Enantiomer sorting has attracted tremendous attention thespinofthelightcanbeperpendiculartothedirectionof – owing to its significant applications in both material science the light beam17 19. -

Chiral Symmetry and Quantum Hadro-Dynamics
Chiral symmetry and quantum hadro-dynamics G. Chanfray1, M. Ericson1;2, P.A.M. Guichon3 1IPNLyon, IN2P3-CNRS et UCB Lyon I, F69622 Villeurbanne Cedex 2Theory division, CERN, CH12111 Geneva 3SPhN/DAPNIA, CEA-Saclay, F91191 Gif sur Yvette Cedex Using the linear sigma model, we study the evolutions of the quark condensate and of the nucleon mass in the nuclear medium. Our formulation of the model allows the inclusion of both pion and scalar-isoscalar degrees of freedom. It guarantees that the low energy theorems and the constrains of chiral perturbation theory are respected. We show how this formalism incorporates quantum hadro-dynamics improved by the pion loops effects. PACS numbers: 24.85.+p 11.30.Rd 12.40.Yx 13.75.Cs 21.30.-x I. INTRODUCTION The influence of the nuclear medium on the spontaneous breaking of chiral symmetry remains an open problem. The amount of symmetry breaking is measured by the quark condensate, which is the expectation value of the quark operator qq. The vacuum value qq(0) satisfies the Gell-mann, Oakes and Renner relation: h i 2m qq(0) = m2 f 2, (1) q h i − π π where mq is the current quark mass, q the quark field, fπ =93MeV the pion decay constant and mπ its mass. In the nuclear medium the quark condensate decreases in magnitude. Indeed the total amount of restoration is governed by a known quantity, the nucleon sigma commutator ΣN which, for any hadron h is defined as: Σ = i h [Q , Q˙ ] h =2m d~x [ h qq(~x) h qq(0) ] , (2) h − h | 5 5 | i q h | | i−h i Z ˙ where Q5 is the axial charge and Q5 its time derivative. -

Tau-Lepton Decay Parameters
58. τ -lepton decay parameters 1 58. τ -Lepton Decay Parameters Updated August 2011 by A. Stahl (RWTH Aachen). The purpose of the measurements of the decay parameters (also known as Michel parameters) of the τ is to determine the structure (spin and chirality) of the current mediating its decays. 58.1. Leptonic Decays: The Michel parameters are extracted from the energy spectrum of the charged daughter lepton ℓ = e, µ in the decays τ ℓνℓντ . Ignoring radiative corrections, neglecting terms 2 →2 of order (mℓ/mτ ) and (mτ /√s) , and setting the neutrino masses to zero, the spectrum in the laboratory frame reads 2 5 dΓ G m = τℓ τ dx 192 π3 × mℓ f0 (x) + ρf1 (x) + η f2 (x) Pτ [ξg1 (x) + ξδg2 (x)] , (58.1) mτ − with 2 3 f0 (x)=2 6 x +4 x −4 2 32 3 2 2 8 3 f1 (x) = +4 x x g1 (x) = +4 x 6 x + x − 9 − 9 − 3 − 3 2 4 16 2 64 3 f2 (x) = 12(1 x) g2 (x) = x + 12 x x . − 9 − 3 − 9 The quantity x is the fractional energy of the daughter lepton ℓ, i.e., x = E /E ℓ ℓ,max ≈ Eℓ/(√s/2) and Pτ is the polarization of the tau leptons. The integrated decay width is given by 2 5 Gτℓ mτ mℓ Γ = 3 1+4 η . (58.2) 192 π mτ The situation is similar to muon decays µ eνeνµ. The generalized matrix element with γ → the couplings gεµ and their relations to the Michel parameters ρ, η, ξ, and δ have been described in the “Note on Muon Decay Parameters.” The Standard Model expectations are 3/4, 0, 1, and 3/4, respectively. -

Chirality-Sorting Optical Forces
Lateral chirality-sorting optical forces Amaury Hayata,b,1, J. P. Balthasar Muellera,1,2, and Federico Capassoa,2 aSchool of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138; and bÉcole Polytechnique, Palaiseau 91120, France Contributed by Federico Capasso, August 31, 2015 (sent for review June 7, 2015) The transverse component of the spin angular momentum of found in Supporting Information. The optical forces arising through evanescent waves gives rise to lateral optical forces on chiral particles, the interaction of light with chiral matter may be intuited by con- which have the unusual property of acting in a direction in which sidering that an electromagnetic wave with helical polarization will there is neither a field gradient nor wave propagation. Because their induce corresponding helical dipole (and multipole) moments in a direction and strength depends on the chiral polarizability of the scatterer. If the scatterer is chiral (one may picture a subwavelength particle, they act as chirality-sorting and may offer a mechanism for metal helix), then the strength of the induced moments depends on passive chirality spectroscopy. The absolute strength of the forces the matching of the helicity of the electromagnetic wave to the also substantially exceeds that of other recently predicted sideways helicity of the scatterer. The forces described in this paper, as well optical forces. as other chirality-sorting optical forces, emerge because the in- duced moments in the scatterer result in radiation in a helicity- optical forces | chirality | optical spin-momentum locking | dependent preferred direction, which causes corresponding recoil. optical spin–orbit interaction | optical spin The effect discussed in this paper enables the tailoring of chirality- sorting optical forces by engineering the local SAM density of optical hirality [from Greek χeιρ (kheir), “hand”] is a type of asym- fields, such that fields with transverse SAM can be used to achieve metry where an object is geometrically distinct from its lateral forces. -

Introduction to Flavour Physics
Introduction to flavour physics Y. Grossman Cornell University, Ithaca, NY 14853, USA Abstract In this set of lectures we cover the very basics of flavour physics. The lec- tures are aimed to be an entry point to the subject of flavour physics. A lot of problems are provided in the hope of making the manuscript a self-study guide. 1 Welcome statement My plan for these lectures is to introduce you to the very basics of flavour physics. After the lectures I hope you will have enough knowledge and, more importantly, enough curiosity, and you will go on and learn more about the subject. These are lecture notes and are not meant to be a review. In the lectures, I try to talk about the basic ideas, hoping to give a clear picture of the physics. Thus many details are omitted, implicit assumptions are made, and no references are given. Yet details are important: after you go over the current lecture notes once or twice, I hope you will feel the need for more. Then it will be the time to turn to the many reviews [1–10] and books [11, 12] on the subject. I try to include many homework problems for the reader to solve, much more than what I gave in the actual lectures. If you would like to learn the material, I think that the problems provided are the way to start. They force you to fully understand the issues and apply your knowledge to new situations. The problems are given at the end of each section. -

Threshold Corrections in Grand Unified Theories
Threshold Corrections in Grand Unified Theories Zur Erlangung des akademischen Grades eines DOKTORS DER NATURWISSENSCHAFTEN von der Fakult¨at f¨ur Physik des Karlsruher Instituts f¨ur Technologie genehmigte DISSERTATION von Dipl.-Phys. Waldemar Martens aus Nowosibirsk Tag der m¨undlichen Pr¨ufung: 8. Juli 2011 Referent: Prof. Dr. M. Steinhauser Korreferent: Prof. Dr. U. Nierste Contents 1. Introduction 1 2. Supersymmetric Grand Unified Theories 5 2.1. The Standard Model and its Limitations . ....... 5 2.2. The Georgi-Glashow SU(5) Model . .... 6 2.3. Supersymmetry (SUSY) .............................. 9 2.4. SupersymmetricGrandUnification . ...... 10 2.4.1. MinimalSupersymmetricSU(5). 11 2.4.2. MissingDoubletModel . 12 2.5. RunningandDecoupling. 12 2.5.1. Renormalization Group Equations . 13 2.5.2. Decoupling of Heavy Particles . 14 2.5.3. One-Loop Decoupling Coefficients for Various GUT Models . 17 2.5.4. One-Loop Decoupling Coefficients for the Matching of the MSSM to the SM ...................................... 18 2.6. ProtonDecay .................................... 21 2.7. Schur’sLemma ................................... 22 3. Supersymmetric GUTs and Gauge Coupling Unification at Three Loops 25 3.1. RunningandDecoupling. 25 3.2. Predictions for MHc from Gauge Coupling Unification . 32 3.2.1. Dependence on the Decoupling Scales µSUSY and µGUT ........ 33 3.2.2. Dependence on the SUSY Spectrum ................... 36 3.2.3. Dependence on the Uncertainty on the Input Parameters ....... 36 3.2.4. Top-Down Approach and the Missing Doublet Model . ...... 39 3.2.5. Comparison with Proton Decay Constraints . ...... 43 3.3. Summary ...................................... 44 4. Field-Theoretical Framework for the Two-Loop Matching Calculation 45 4.1. TheLagrangian.................................. 45 4.1.1. -

The Taste of New Physics: Flavour Violation from Tev-Scale Phenomenology to Grand Unification Björn Herrmann
The taste of new physics: Flavour violation from TeV-scale phenomenology to Grand Unification Björn Herrmann To cite this version: Björn Herrmann. The taste of new physics: Flavour violation from TeV-scale phenomenology to Grand Unification. High Energy Physics - Phenomenology [hep-ph]. Communauté Université Grenoble Alpes, 2019. tel-02181811 HAL Id: tel-02181811 https://tel.archives-ouvertes.fr/tel-02181811 Submitted on 12 Jul 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The taste of new physics: Flavour violation from TeV-scale phenomenology to Grand Unification Habilitation thesis presented by Dr. BJÖRN HERRMANN Laboratoire d’Annecy-le-Vieux de Physique Théorique Communauté Université Grenoble Alpes Université Savoie Mont Blanc – CNRS and publicly defended on JUNE 12, 2019 before the examination committee composed of Dr. GENEVIÈVE BÉLANGER CNRS Annecy President Dr. SACHA DAVIDSON CNRS Montpellier Examiner Prof. ALDO DEANDREA Univ. Lyon Referee Prof. ULRICH ELLWANGER Univ. Paris-Saclay Referee Dr. SABINE KRAML CNRS Grenoble Examiner Prof. FABIO MALTONI Univ. Catholique de Louvain Referee July 12, 2019 ii “We shall not cease from exploration, and the end of all our exploring will be to arrive where we started and know the place for the first time.” T. -
CHIRALITY •The Helicity Eigenstates for a Particle/Anti-Particle for Are
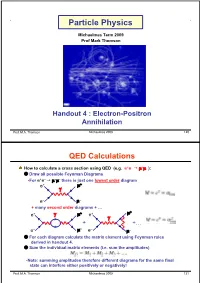
Particle Physics Michaelmas Term 2009 Prof Mark Thomson Handout 4 : Electron-Positron Annihilation Prof. M.A. Thomson Michaelmas 2009 120 QED Calculations How to calculate a cross section using QED (e.g. e+e– $ +– ): Draw all possible Feynman Diagrams •For e+e– $ +– there is just one lowest order diagram + e e– – + many second order diagrams + … + + e e + +… e– – e– – For each diagram calculate the matrix element using Feynman rules derived in handout 4. Sum the individual matrix elements (i.e. sum the amplitudes) •Note: summing amplitudes therefore different diagrams for the same final state can interfere either positively or negatively! Prof. M.A. Thomson Michaelmas 2009 121 and then square this gives the full perturbation expansion in • For QED the lowest order diagram dominates and for most purposes it is sufficient to neglect higher order diagrams. + + e e e– – e– – Calculate decay rate/cross section using formulae from handout 1. •e.g. for a decay •For scattering in the centre-of-mass frame (1) •For scattering in lab. frame (neglecting mass of scattered particle) Prof. M.A. Thomson Michaelmas 2009 122 Electron Positron Annihilation ,Consider the process: e+e– $ +– – •Work in C.o.M. frame (this is appropriate – + for most e+e– colliders). e e •Only consider the lowest order Feynman diagram: + ( Feynman rules give: e •Incoming anti-particle – – NOTE: e •Incoming particle •Adjoint spinor written first •In the C.o.M. frame have with Prof. M.A. Thomson Michaelmas 2009 123 Electron and Muon Currents •Here and matrix element • In handout 2 introduced the four-vector current which has same form as the two terms in [ ] in the matrix element • The matrix element can be written in terms of the electron and muon currents and • Matrix element is a four-vector scalar product – confirming it is Lorentz Invariant Prof. -

Neutrino Masses-How to Add Them to the Standard Model
he Oscillating Neutrino The Oscillating Neutrino of spatial coordinates) has the property of interchanging the two states eR and eL. Neutrino Masses What about the neutrino? The right-handed neutrino has never been observed, How to add them to the Standard Model and it is not known whether that particle state and the left-handed antineutrino c exist. In the Standard Model, the field ne , which would create those states, is not Stuart Raby and Richard Slansky included. Instead, the neutrino is associated with only two types of ripples (particle states) and is defined by a single field ne: n annihilates a left-handed electron neutrino n or creates a right-handed he Standard Model includes a set of particles—the quarks and leptons e eL electron antineutrino n . —and their interactions. The quarks and leptons are spin-1/2 particles, or weR fermions. They fall into three families that differ only in the masses of the T The left-handed electron neutrino has fermion number N = +1, and the right- member particles. The origin of those masses is one of the greatest unsolved handed electron antineutrino has fermion number N = 21. This description of the mysteries of particle physics. The greatest success of the Standard Model is the neutrino is not invariant under the parity operation. Parity interchanges left-handed description of the forces of nature in terms of local symmetries. The three families and right-handed particles, but we just said that, in the Standard Model, the right- of quarks and leptons transform identically under these local symmetries, and thus handed neutrino does not exist. -

Chirality and the Angular Momentum of Light
View metadata, citation andDownloaded similar papers from at http://rsta.royalsocietypublishing.org/core.ac.uk on February 21, 2017 brought to you by CORE Chirality and the angular provided by Enlighten momentum of light rsta.royalsocietypublishing.org Robert P.Cameron1,JörgB.Götte1,†, Stephen M. Barnett1 andAlisonM.Yao2 Research 1School of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, UK Cite this article: Cameron RP,Götte JB, 2Department of Physics, University of Strathclyde, Barnett SM, Yao AM. 2017 Chirality and the GlasgowG40NG,UK angular momentum of light. Phil.Trans.R. RPC, 0000-0002-8809-5459 Soc. A 375: 20150433. http://dx.doi.org/10.1098/rsta.2015.0433 Chirality is exhibited by objects that cannot be rotated into their mirror images. It is far from obvious that Accepted: 12 August 2016 this has anything to do with the angular momentum of light, which owes its existence to rotational symmetries. There is nevertheless a subtle connection One contribution of 14 to a theme issue between chirality and the angular momentum of light. ‘Optical orbital angular momentum’. We demonstrate this connection and, in particular, its significance in the context of chiral light–matter Subject Areas: interactions. atomic and molecular physics, optics This article is part of the themed issue ‘Optical orbital angular momentum’. Keywords: chirality, optical angular momentum, molecules, physical chemistry 1. Introduction Author for correspondence: The word chiral was introduced by Kelvin to refer to any geometrical figure or group of points that Robert P.Cameron cannot be brought into coincidence with its mirror e-mail: [email protected] image, thus possessing a sense of handedness [1]. -

Chiral Symmetry and Lattice Fermions
Chiral Symmetry and Lattice Fermions David B. Kaplan Institute for Nuclear Theory, University of Washington, Seattle, WA 98195-1550 Lectures delivered at the Les Houches Ecole´ d'Et´ede´ Physique Th´eorique arXiv:0912.2560v2 [hep-lat] 18 Jan 2012 Acknowledgements I have benefitted enormously in my understanding of lattice fermions from conversa- tions with Maarten Golterman, Pilar Hernandez, Yoshio Kikukawa, Martin L¨uscher, Rajamani Narayanan, Herbert Neuberger, and Stephen Sharpe. Many thanks to them (and no blame for the content of these notes). This work is supported in part by U.S. DOE grant No. DE-FG02-00ER41132. Contents 1 Chiral symmetry 1 1.1 Introduction 1 1.2 Spinor representations of the Lorentz group 1 1.3 Chirality in even dimensions 3 1.4 Chiral symmetry and fermion mass in four dimensions 4 1.5 Weyl fermions 6 1.6 Chiral symmetry and mass renormalization 9 1.7 Chiral symmetry in QCD 11 1.8 Fermion determinants in Euclidian space 13 1.9 Parity and fermion mass in odd dimensions 15 1.10 Fermion masses and regulators 15 2 Anomalies 17 2.1 The U(1)A anomaly in 1+1 dimensions 17 2.2 Anomalies in 3+1 dimensions 20 2.3 Strongly coupled chiral gauge theories 24 2.4 The non-decoupling of parity violation in odd dimensions 25 3 Domain Wall Fermions 27 3.1 Chirality, anomalies and fermion doubling 27 3.2 Domain wall fermions in the continuum 28 3.3 Domain wall fermions and the Callan-Harvey mechanism 31 3.4 Domain wall fermions on the lattice 35 4 Overlap fermions and the Ginsparg-Wilson equation 40 4.1 Overlap fermions 40 4.2 The Ginsparg-Wilson equation and its consequences 44 4.3 Chiral gauge theories: the challenge 48 References 49 1 Chiral symmetry 1.1 Introduction Chiral symmetries play an important role in the spectrum and phenomenology of both the standard model and various theories for physics beyond the standard model. -

Cosmological Chirality and Magnetic Fields from Parity Violating Particle
Cosmological chirality and magnetic fields from parity violating particle decays Tanmay Vachaspati∗, Alexander Vilenkin† ∗Physics Department, Arizona State University, Tempe, AZ 85287, USA. †Institute of Cosmology, Department of Physics and Astronomy, Tufts University, Medford, MA 02155, USA. We estimate the chirality of the cosmological medium due to parity violating decays of standard model particles, focusing on the example of tau leptons. The non-trivial chirality is however too small to make a significant contribution to the cosmological magnetic field via the chiral-magnetic effect. I. INTRODUCTION summarized and discussed in Sec. IV. The last few decades have seen growing interest in cos- mic magnetic fields on several fronts [1]. Several ideas II. CHIRALITY PRODUCTION IN TAU have been proposed that can generate magnetic fields in DECAYS cosmology, some of which are directly tied to known par- ticle physics [2–4] and its possible extensions [1, 5–8]. To illustrate the physics of the effect, in this section we The magneto-hydrodynamical (MHD) evolution of cos- will discuss the decay of tau leptons in the background mological magnetic fields is now understood quite well of a magnetic field and fluid vorticity. Except for small on the basis of analytical arguments [9, 10] and direct differences, the physics carries over to the case of decays simulations [11]. There are claims for an indirect lower of other particles. bound on the cosmological magnetic field strength [12– 16], though not without debate [17, 18], and more direct evidence [19]. Concurrently there are claims of a par- A. Particle decay ity violating signature that can be used to measure the magnetic field helicity spectrum [20, 21] though with no Tau leptons decay into electrons (or muons) and neu- significant detections as yet [22, 23].