Partial Tetrasomy of Chromosome 22Q11.1 Resulting from a Supernumerary Isodicentric Marker Chromosome in a Boy with Cat-Eye Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Role of RUNX1 in Aberrant Retinal Angiogenesis Jonathan D
Page 1 of 25 Diabetes Identification of RUNX1 as a mediator of aberrant retinal angiogenesis Short Title: Role of RUNX1 in aberrant retinal angiogenesis Jonathan D. Lam,†1 Daniel J. Oh,†1 Lindsay L. Wong,1 Dhanesh Amarnani,1 Cindy Park- Windhol,1 Angie V. Sanchez,1 Jonathan Cardona-Velez,1,2 Declan McGuone,3 Anat O. Stemmer- Rachamimov,3 Dean Eliott,4 Diane R. Bielenberg,5 Tave van Zyl,4 Lishuang Shen,1 Xiaowu Gai,6 Patricia A. D’Amore*,1,7 Leo A. Kim*,1,4 Joseph F. Arboleda-Velasquez*1 Author affiliations: 1Schepens Eye Research Institute/Massachusetts Eye and Ear, Department of Ophthalmology, Harvard Medical School, 20 Staniford St., Boston, MA 02114 2Universidad Pontificia Bolivariana, Medellin, Colombia, #68- a, Cq. 1 #68305, Medellín, Antioquia, Colombia 3C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, 55 Fruit St., Boston, MA 02114 4Retina Service, Massachusetts Eye and Ear Infirmary, Department of Ophthalmology, Harvard Medical School, 243 Charles St., Boston, MA 02114 5Vascular Biology Program, Boston Children’s Hospital, Department of Surgery, Harvard Medical School, 300 Longwood Ave., Boston, MA 02115 6Center for Personalized Medicine, Children’s Hospital Los Angeles, Los Angeles, 4650 Sunset Blvd, Los Angeles, CA 90027, USA 7Department of Pathology, Harvard Medical School, 25 Shattuck St., Boston, MA 02115 Corresponding authors: Joseph F. Arboleda-Velasquez: [email protected] Ph: (617) 912-2517 Leo Kim: [email protected] Ph: (617) 912-2562 Patricia D’Amore: [email protected] Ph: (617) 912-2559 Fax: (617) 912-0128 20 Staniford St. Boston MA, 02114 † These authors contributed equally to this manuscript Word Count: 1905 Tables and Figures: 4 Diabetes Publish Ahead of Print, published online April 11, 2017 Diabetes Page 2 of 25 Abstract Proliferative diabetic retinopathy (PDR) is a common cause of blindness in the developed world’s working adult population, and affects those with type 1 and type 2 diabetes mellitus. -

(OKCD03366) Lot# KC2621
ADA2 Chemi-Luminescent ELISA Kit (Human) (OKCD03366) Lot# KC2621 Instructions for use For the quantitative measurement of ADA2 in serum, plasma, tissue homogenates, cell lysates, cell culture supernatants and other biological fluids. Variation between lots can occur. Refer to the manual provided with the kit. This product is intended for research use only. ADA2 Chemi-Luminescent ELISA Kit (Human) (OKCD03366) – Lot# KC2621 Table of Contents 1. Background ............................................................................................................................................. 2 2. Assay Summary ..................................................................................................................................... 3 3. Storage and Stability .............................................................................................................................. 3 4. Kit Components ...................................................................................................................................... 3 5. Precautions ............................................................................................................................................. 4 6. Required Materials Not Supplied ......................................................................................................... 4 7. Technical Application Tips .................................................................................................................... 4 8. Reagent Preparation ............................................................................................................................. -

Supplementary Figures and Tables
SUPPLEMENTARY DATA Supplementary Figure 1. Isolation and culture of endothelial cells from surgical specimens of FVM. (A) Representative pre-surgical fundus photograph of a right eye exhibiting a FVM encroaching on the optic nerve (dashed line) causing tractional retinal detachment with blot hemorrhages throughout retina (arrow heads). (B) Magnetic beads (arrows) allow for separation and culturing of enriched cell populations from surgical specimens (scale bar = 100 μm). (C) Cultures of isolated cells stained positively for CD31 representing a successfully isolated enriched population (scale bar = 40 μm). ©2017 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db16-1035/-/DC1 SUPPLEMENTARY DATA Supplementary Figure 2. Efficient siRNA knockdown of RUNX1 expression and function demonstrated by qRT-PCR, Western Blot, and scratch assay. (A) RUNX1 siRNA induced a 60% reduction of RUNX1 expression measured by qRT-PCR 48 hrs post-transfection whereas expression of RUNX2 and RUNX3, the two other mammalian RUNX orthologues, showed no significant changes, indicating specificity of our siRNA. Functional inhibition of Runx1 signaling was demonstrated by a 330% increase in insulin-like growth factor binding protein-3 (IGFBP3) RNA expression level, a known target of RUNX1 inhibition. Western blot demonstrated similar reduction in protein levels. (B) siRNA- 2’s effect on RUNX1 was validated by qRT-PCR and western blot, demonstrating a similar reduction in both RNA and protein. Scratch assay demonstrates functional inhibition of RUNX1 by siRNA-2. ns: not significant, * p < 0.05, *** p < 0.001 ©2017 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db16-1035/-/DC1 SUPPLEMENTARY DATA Supplementary Table 1. -

Plasma Cells in Vitro Generation of Long-Lived Human
Downloaded from http://www.jimmunol.org/ by guest on September 24, 2021 is online at: average * The Journal of Immunology , 32 of which you can access for free at: 2012; 189:5773-5785; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication November 2012; doi: 10.4049/jimmunol.1103720 http://www.jimmunol.org/content/189/12/5773 In Vitro Generation of Long-lived Human Plasma Cells Mario Cocco, Sophie Stephenson, Matthew A. Care, Darren Newton, Nicholas A. Barnes, Adam Davison, Andy Rawstron, David R. Westhead, Gina M. Doody and Reuben M. Tooze J Immunol cites 65 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/11/16/jimmunol.110372 0.DC1 This article http://www.jimmunol.org/content/189/12/5773.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 24, 2021. The Journal of Immunology In Vitro Generation of Long-lived Human Plasma Cells Mario Cocco,*,1 Sophie Stephenson,*,1 Matthew A. -
![Bs-9005R-Cy7 [Conjugated Primary Antibody]](https://docslib.b-cdn.net/cover/2092/bs-9005r-cy7-conjugated-primary-antibody-1182092.webp)
Bs-9005R-Cy7 [Conjugated Primary Antibody]
bs-9005R-Cy7 [Conjugated Primary Antibody] Rabbit Anti-CECR1 Polyclonal Antibody, Cy7 Conjugated www.biossusa.com [email protected] 800.501.7654 [DOMESTIC] +1.781.569.5821 [INTERNATIONAL] D A T A S H E E T Host: Rabbit Conjugation: Cy7 Target Protein: CECR1 Excitation/ 743nm/767nm Emission: Clonality: Polyclonal Isotype: IgG Size: 100ul Entrez Gene: 51816 Concentration: 1ug/ul Swiss Prot: Applications: IF(IHC-P)(1:50-200) Source: KLH conjugated synthetic peptide derived from human CECR1/ADGF Cross Reactive Human Species: Purification: Purified by Protein A. Caution: For research use only. Not for human or animal Storage: Aqueous buffered solution containing 100ug/ml BSA, 50% glycerol and 0.09% sodium azide. therapeutic or diagnostic use. Store at 4°C for 12 months. Background: CECR1 is a member of the adenosine and AMP deaminases family. It may act as a growth factor and have adenosine deaminase activity. It is a candidate gene for cat eye syndrome. Two transcript variants encoding distinct isoforms have been identified for this gene.Adenosine deaminase is an enzyme that is present in most tissues and exists predominantly as a monomer, although in some tissues it is associated with adenosine deaminase-binding protein. Adenosine deaminase degrades extracellular adenosine, which is toxic for lymphocytes. A novel family of growth factors that share sequence similarity to adenosine deaminase has been identified. The cat eye syndrome critical region protein (CECR) family includes CECR1, CECR2, CECR3, CECR4, CECR5, CECR6, CECR7, CECR8 and CECR9. The genes encoding CECR proteins are candidates for Cat Eye Syndrome (CES), a developmental disorder associated with the duplication of a 2 Mb region of 22q11.2. -

Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis
Getting “Under the Skin”: Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis by Kristen Monét Brown A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Epidemiological Science) in the University of Michigan 2017 Doctoral Committee: Professor Ana V. Diez-Roux, Co-Chair, Drexel University Professor Sharon R. Kardia, Co-Chair Professor Bhramar Mukherjee Assistant Professor Belinda Needham Assistant Professor Jennifer A. Smith © Kristen Monét Brown, 2017 [email protected] ORCID iD: 0000-0002-9955-0568 Dedication I dedicate this dissertation to my grandmother, Gertrude Delores Hampton. Nanny, no one wanted to see me become “Dr. Brown” more than you. I know that you are standing over the bannister of heaven smiling and beaming with pride. I love you more than my words could ever fully express. ii Acknowledgements First, I give honor to God, who is the head of my life. Truly, without Him, none of this would be possible. Countless times throughout this doctoral journey I have relied my favorite scripture, “And we know that all things work together for good, to them that love God, to them who are called according to His purpose (Romans 8:28).” Secondly, I acknowledge my parents, James and Marilyn Brown. From an early age, you two instilled in me the value of education and have been my biggest cheerleaders throughout my entire life. I thank you for your unconditional love, encouragement, sacrifices, and support. I would not be here today without you. I truly thank God that out of the all of the people in the world that He could have chosen to be my parents, that He chose the two of you. -

Characterized by Array CGH in a Boy with Cat-Eye Syndrome Irén Haltrich1*, Henriett Pikó2, Eszter Kiss1, Zsuzsa Tóth1, Veronika Karcagi2 and György Fekete1
Haltrich et al. Molecular Cytogenetics 2014, 7:37 http://www.molecularcytogenetics.org/content/7/1/37 CASE REPORT Open Access A de novo atypical ring sSMC(22) characterized by array CGH in a boy with cat-eye syndrome Irén Haltrich1*, Henriett Pikó2, Eszter Kiss1, Zsuzsa Tóth1, Veronika Karcagi2 and György Fekete1 Abstract Background: Microduplications 22q11 have been characterized as a genomic duplication syndrome mediated by nonallelic homologous recombination between region-specific low-copy repeats. Here we report on a 19 years old boy with intellectual disability having an unexpected structurally complex ring small supernumerary marker chromosome (sSMC) originated from a larger trisomy and a smaller tetrasomy of proximal 22q11 harboring additional copies of cat eye syndrome critical regions genes. Results: Principal clinical features were: anorectal and urogenital malformations, total anomalous pulmonary venous return with secundum ASD, hearing defect, preauricular pits, seizure and eczema. The proband also presented some rare or so far not reported clinical findings such as hyperinsulinaemia, severe immunodeficiency and grave cognitive deficits. Chromosome analysis revealed a mosaic karyotype with the presence of a small ring-like marker in 60% of cells. Array CGH detected approximately an 1,2 Mb single and a 0,2 Mb double copy gain of the proximal long arm of chromosome 22. The 1,3 Mb intervening region of chromosome 22 from centromere to the breakpoints showed no copy alteration. The karyotype of the patient was defined as 47,XY,+mar[60]/46,XY[40].ish idic r(22)(q11.1.q11.21) × 4.arr 22q11(17,435, 645-18,656,678) × 3,(17,598,642-17,799,783) × 4 dn. -

Identification of Novel Genetic Mutations Leading to Rare Monogenic Inflammatory Diseases
University College London Identification of novel genetic mutations leading to rare monogenic inflammatory diseases Ciara Maria Mulhern Thesis submitted for PhD Infection, Immunity and Inflammation Research and Teaching Department 1 UCL, Great Ormond Street, Institute of Child Health I, Ciara Maria Mulhern, state that all experimental and analytical work presented in this thesis has been carried out by myself. Where others have contributed to this work, this has been indicated in the thesis. 2 Acknowledgements Firstly, I would like to say a huge thank you to my supervisors; Dr Despina Eleftheriou, Dr Ying Hong and Professor Paul Brogan. I cannot say enough words of thanks to Despina and Ying, two incredible women. Despina has always been so supportive and encouraging. Thank you for always being consistently present throughout my PhD, even when you were on holidays, you still answered my emails. No problem is ever too small for you. Ying, Thank you for always advising me, pushing me and giving me confidence in my work. Thank you for showing me how to work as a proper scientist, how to carry out assays, plan experiments and most importantly, how to get assays to work. You have been a constant support throughout my time at ICH, not just to me but to everyone around you. Thank you as well to Paul; your constant optimism and positivity was always appreciated. Thank you for your enthusiasm and words of encouragement. I would also like to thank Ebun and Dara two mighty postdocs. Ebun, you are a constant delight, always smiling and happy. Thank you for your expertise while gene hunting; you are an absolute pleasure to work with. -

1 MAP3K6 Mutations in a Neurovascular Disease Causing
MAP3K6 mutations in a neurovascular disease causing stroke, cognitive impairment and tremor Authors: Andreea Ilinca, Elisabet Englund, Sofie Samuelsson, Katarina Truvé, Efthymia Kafantari, Nicolas Martinez-Majander, Jukka Putaala, Claes Håkansson, Arne G. Lindgren, Andreas Puschmann Supplemental data: table e1: Detailed clinical information on the studied individuals ............................................ 2 supplemental data e1: Genetic examinations of the proband and family ................................ 4 supplemental data e1 – table 1 .........................................................................................................4 supplemental data e1 – table 2 .........................................................................................................6 Detailed description of the bioinformatics pipeline used for WGS analyses ............................7 Co-segregation analyses of WES data from eight family members ...........................................8 supplemental data e1 – table 3 .........................................................................................................8 supplemental data e1 – table 4 .........................................................................................................8 supplemental data e1 – table 5 .........................................................................................................9 Determining the shared haplotype around MAP3K6 c.322G>A in affected members ......... 10 Analysis of sequence alterations, short tandem repeats -
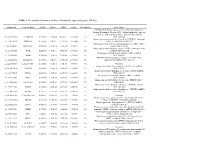
Table 1 the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table 1 The statistical metrics for key differentially expressed genes (DEGs) Agiliant Id Gene Symbol logFC pValue FDR tvalue Regulation Gene Name PREDICTED: Homo sapiens similar to Filamin-C (Gamma- filamin) (Filamin-2) (Protein FLNc) (Actin-binding-like protein) (ABP-L) (ABP-280-like protein) (LOC649056), mRNA A_24_P237896 LOC649056 0.509843 9.18E-14 4.54E-11 12.07302 Up [XR_018580] Homo sapiens programmed cell death 10 (PDCD10), transcript A_23_P18325 PDCD10 0.243111 2.8E-12 9.24E-10 10.62808 Up variant 1, mRNA [NM_007217] Homo sapiens Rho GTPase activating protein 27 (ARHGAP27), A_23_P141335 ARHGAP27 0.492709 3.97E-12 1.22E-09 10.48571 Up mRNA [NM_199282] Homo sapiens tubby homolog (mouse) (TUB), transcript variant A_23_P53110 TUB 0.528219 1.77E-11 4.56E-09 9.891033 Up 1, mRNA [NM_003320] Homo sapiens MyoD family inhibitor (MDFI), mRNA A_23_P42168 MDFI 0.314474 1.81E-10 3.74E-08 8.998697 Up [NM_005586] PREDICTED: Homo sapiens hypothetical LOC644701 A_32_P56890 LOC644701 0.444703 3.6E-10 7.09E-08 8.743973 Up (LOC644701), mRNA [XM_932316] A_32_P167111 A_32_P167111 0.873588 7.41E-10 1.4E-07 8.47781 Up Unknown Homo sapiens zinc finger protein 784 (ZNF784), mRNA A_24_P221424 ZNF784 0.686781 9.18E-10 1.68E-07 8.399687 Up [NM_203374] Homo sapiens lin-28 homolog (C. elegans) (LIN28), mRNA A_23_P74895 LIN28 0.218876 1.27E-09 2.24E-07 8.282224 Up [NM_024674] Homo sapiens ribosomal protein L5 (RPL5), mRNA A_23_P12140 RPL5 0.247598 1.81E-09 3.11E-07 8.154317 Up [NM_000969] Homo sapiens cDNA FLJ43841 fis, clone TESTI4006137. -

Cecr2 Mutant Mice As a Model for Human Cat Eye Syndrome Renée Dicipulo1, Kacie A
www.nature.com/scientificreports OPEN Cecr2 mutant mice as a model for human cat eye syndrome Renée Dicipulo1, Kacie A. Norton1, Nicholas A. Fairbridge1,2, Yana Kibalnyk1, Sabrina C. Fox1, Lisa K. Hornberger3 & Heather E. McDermid1* Cat eye syndrome (CES), a human genetic disorder caused by the inverted duplication of a region on chromosome 22, has been known since the late 1890s. Despite the signifcant impact this disorder has on afected individuals, models for CES have not been produced due to the difculty of efectively duplicating the corresponding chromosome region in an animal model. However, the study of phenotypes associated with individual genes in this region such as CECR2 may shed light on the etiology of CES. In this study we have shown that deleterious loss of function mutations in mouse Cecr2 efectively demonstrate many of the abnormal features present in human patients with CES, including coloboma and specifc skeletal, kidney and heart defects. Beyond phenotypic analyses we have demonstrated the importance of utilizing multiple genetic backgrounds to study disease models, as we see major diferences in penetrance of Cecr2-related abnormal phenotype between mouse strains, reminiscent of the variability in the human syndrome. These fndings suggest that Cecr2 is involved in the abnormal features of CES and that Cecr2 mice can be used as a model system to study the wide range of phenotypes present in CES. Cat eye syndrome (CES) is a rare human chromosomal disorder characterized by a highly variable phenotype 1. Te name comes from the eye defect coloboma, which results from the failure of the optic fssure to close during early eye development. -

US 2017/0166878 A9 (12) Patent Application Publication (48) Pub
US 20170166878A9 (19) United States (10) Pub. No.: US 2017/0166878 A9 (12) Patent Application Publication (48) Pub. Date: Jun. 15, 2017 Thanos et al. CORRECTED PUBLICATION (54) COMPOSITIONS OF ADENOSINE (60) Provisional application No. 62/063.936, filed on Oct. DEAMINASE-2 (ADA2), VARIANTS 14, 2014. THEREOF AND METHODS OF USING SAME (71) Applicant: Halozyme, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Christopher D. Thanos, Tiburon, CA (51) Int. Cl. (US); Lin Wang, San Diego, CA (US); CI2N 9/78 (2006.01) H. Michael Shepard, San Diego, CA A638/48 (2006.01) (US) (52) U.S. Cl. (21) Appl. No.: 15/094,908 CPC ................ CI2N 9/78 (2013.01); A61K 38/48 (22) Filed1C Apr.pr. 8,5, 2016 (2013.01); C12Y 305/04004 (2013.01) Prior Publication Data (57) ABSTRACT (15) Correction of US 2016/0272960 A1 Sep. 22, 2016 See Claim 11. Provided are variant adenosine deaminase 2 (ADA2) pro teins, conjugates thereof and compositions containing the (65) US 2016/0272960 A1 Sep. 22, 2016 proteins and/or conjugates. Also provided are methods and uses of the ADA2 proteins or conjugates for treating dis Related U.S. Application Data eases and conditions, such as a tumor or cancer, and in (63) Continuation-in-part of application No. PCT/US 15/ particular any disease or condition associated with elevated 55613, filed on Oct. 14, 2015. adenosine or other associated marker. Patent Application Publication Jun. 15, 2017. Sheet 2 of 7 US 2017/O166878 A9 09 09 OZI OZT 08T 08T 0€ 08 099 O9€. NVITT ?VÄTHOEIXIT,N'HA AVIACIXIS AVIAQIXIS (ISA'I>{'IAÕNSHADAHIdI (ISATXHTAONSTOEOATRIÐI × * * ANCIÐARGÖWISHHA?CIH,BA?IVÄHITIÐSILAH,HINE,EXISMAANÕNLÄIAETHÕLATILHNHITIS ×* ?THINKIXEXTTTHTHIEGIST,SÐHÆSINGAVITIHOTw???s?ÐGAT? ANCI?ZJ?ÕWISHRAZG?JA?V?KHITIÐSILAH,HIJ,GI,HXISMAANÕNL?CIATHAHÕJATI,HÌNHTITIS }MSKIV?HXISTV7H?H?IHLLNTW'IVOITIINHCIISLÐÕMCI MS?L?AVGIHXISTIVZH?H?INILINTW'IVOITINNIGISL5)ÕMC? ILGALVACIWIEXCIMEXXIMI?WHLNXESETTLS?, eTT?Ios) eTTTIORÐ e[|LIOÐ eTT?LIOÐ Ueu?rlH UelunH.