A Comparative Study of Solvent Extraction, Soxhlet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hydro-Distillation and Steam Distillation from Aromatic Plants
Hydro-distillation and steam distillation from aromatic plants Sudeep Tandon Scientist Chemical Engineering Division CIMAP, Lucknow HISTORY Written records of herbal distillation are found as early as the first century A.D., and around 1000 A.D., the noted Arab physician and naturalist Ibn Sina also known as Avicenna described the distillation of rose oil from rose petals The ancient Arabian people began to study the chemical properties of essential oils & developed and refined the distillation process Europeans began producing essential oils in the 12th century 1 DISTILLATION ? A process in which a liquid or vapour mixture of two or more substances is separated into its component fractions of desired purity, by the application and removal of heat. In simple terms distillation of aromatic herbs implies vaporizing or liberating the oils from the trichomes / plant cell membranes of the herb in presence of high temperature and moisture and then cooling the vapour mixture to separate out the oil from water. It is the most popular widely used and cost effective method in use today for producing majority of the essential oils throughout the world Distillation is an art and not just a “Chemical" process that is reliant upon many factors for successful quality oil production. BASIC SCIENTIFIC PRINCIPLES INVOLVED IN THE PROCESS To convert any liquid into a vapour we have to apply energy in form of heat called as latent heat of vaporization A liquid always boils at the temperature at which its vapour pressure equals the atmospheric / surrounding pressure For two immiscible liquids the total vapour pressure of the mixture is always equal to the sum of their partial pressures The composition of the mixture will be determined by the concentration of the individual components into its partial pressure As known the boiling point of most essential oil components exceeds that of water and generally lies between 150 – 300oC 2 If a sample of an essential oil having a component ‘A’ having boiling point for example 190oC and the boiling point of the water is 100oC. -

Supercritical CO Extraction of Essential Oil from Clove
Supercritical CO2 Extraction of Essential Oil from Clove Bud: Effect of Operation Conditions on the Selective Isolation of Eugenol and Eugenyl Acetate Farshad Yazdani, Morteza Mafi, Fathollah Farhadi, Kourosh Tabar-Heidar, Kioumars Aghapoor, Farshid Mohsenzadeh, and Hossein Reza Darabi Chemistry & Chemical Engineering Research Center of Iran, P.O. Box 14335-186, Teheran, Iran Reprint requests to Dr. H. R. Darabi. E-mail: [email protected] Z. Naturforsch. 60b, 1197 – 1201 (2005); received July 14, 2005 The supercritical fluid extraction (SFE) of clove bud essential oil was studied using CO2 as sol- vent. The effect of operation conditions was analyzed in a series of experiments at temperatures between 325 and 416 K and pressures between 110 and 190 bar. The collected extracts were ana- lyzed and the relative composition of the essential oil was determined. The optimum condition was found in a temperature of 353 K and at a pressure of 190 bar, minimizing the number of extracts to two compounds (eugenol and eugenyl acetate). The extract obtained from clove bud by using su- percritical fluid extraction was compared with the essential oil obtained by steam distillation and microwave-assisted extraction by considering both quantity and quality of the product. The oil yield was higher in steam distillation and microwave oven extraction. In contrast, oil extracted by using SFE contained higher amount of eugenol and eugenyl acetate. Key words: Clove Bud, Eugenol, Extraction Introduction The high pressure is used for processes where to- Identification, extraction and preparation of clove tal extraction of a target compound is desired, since bud oil are gaining interest as its applications are most extractable compounds exhibit their maximum becoming widespread in different sectors like food, solubility in a supercritical solvent at higher pressures. -

The Field Distillation of Peppermint Oil
:__ / C. OREGON COLLLCTlop Improvements in The Field Distillation of Peppermint Oil By A. D. HUGHES Professor of Mechanical Engineering Bulletin No. 31 August 1952 A Cooperative Research Project of the Engineering Experiment Station and the Agricultural Experiment Station. Engineering Experiment Station Oregon State College Corvallis THE Oregon State Engineering Experiment Station was established by act of the Board of Regents of Oregon State College on May 4, 1927. It is the purpose of the Station to serve the state in a manner broadly outlined by the following policy: (1)To stimulate and elevate engineering education by developing the research spirit in faculty and students. (2) To serve the industries,utilities, professional engi- neers, public departments, and engineering teachers by making investigations of interest to them. (3) To publish and distribute by bulletins, circulars, and technical articles in periodicals the results of such studies, sur- veys, tests, investigations, and research as will be of greatest benefit to the people of Oregon, and particularly to the state's industries, utilities, and professional engineers. To make available the results of the investigations con- ducted by the Station three types of publications are issued. These are: (1) Bulletins covering original investigations. (2) Circulars giving compilations of useful data (3) Reprints giving more general distribution to scientific papers or reports previously published elsewhere, as for exam- ple, in the proceedings of professional societies. Single copies of publications are sent free on request to residents of Oregon, to libraries, and to other experiment sta- tions exchanging publications.As long as available, additional copies, or copies to others, are sent at prices covering cost of printing. -

Part I. Isolation of (R)-(+)-Limonene from Grapefruit Or Orange Peel
EXPERIMENT 5 ISOLATION OF LIMONENE FROM NATURAL PRODUCTS: STEAM DISTILLATION AND SOXHLET EXTRACTION OF CITRUS PEELS & MINT LEAVES Part 1: Isolation of limonene from citrus peel by steam distillation Part 2: Isolation of limonene from mint leaves by soxhlet extraction Students will work in teams of two. Each team will do one steam distillation, and one soxhlet extraction. If there is an odd number of students, then the student will do either a steam distillation or extraction, depending on availability of glassware. Reading Assignment: Pavia, Sections 18.1-18.4, 12.12 Pre-lab Questions: 1) Questions #1, 2, Pavia p. 284 - 285 2) In the spearmint leaf extraction, indicate which of the following solvents may or may not be suitable as a replacement for the hexane used in the experiment: acetone, diethyl ether, ethanol. Briefly explain your choices. Part I. Isolation of (R)-(+)-Limonene from Grapefruit or Orange Peel: (R)-(+)-limonene Line and 3-D structures of (R)-(+)-limonene. The major constituent of the steam-volatile oil of grapefruit or orange peel is (R)-(+)-limonene. It can be isolated as an esential oil of about 97% purity by a steam distillation of citrus fruit peels. Attempting to distill this essential oil directly from plant CHEM M52LA/H52LA Experiment 5 Page 1 763 material is generally not feasible. Limonene is a high-boiling liquid (bp = 175.5-176 °C) which decomposes under the high heat needed to bring it to a boil. Steam distillation is a much gentler way to obtain essential oils, as it allows the distillation of co-mixture of oil and water at a boiling point less than 100 °C. -

Recent Advances on Supercritical Fluid Extraction of Essential Oils
African Journal of Pharmacy and Pharmacology Vol. 5(9), pp. 1196-1211, 8 September, 2011 Available online http://www.academicjournals.org/ajpp DOI: 10.5897/AJPP11.228 ISSN 1996-0816 ©2011 Academic Journals Full Length Research Paper Recent advances on supercritical fluid extraction of essential oils Lili Xu1, Xiaori Zhan1*, Zhaowu Zeng2*, Rong Chen1, Haifeng Li1, Tian Xie1 and Shuling Wang1 1Research Center for Biomedicine and Health, Hangzhou Normal University, 1378 Wen Yi Xi Road, Hangzhou, Zhejiang, 311121, People’s Republic of China. 2Guangdong Key Laboratory for Research and Development of Natural Drugs, Guangdong Medical College, Dongguan, Guangdong, 523808, People’s Republic of China. Accepted 2 August, 2011 Supercritical fluid extraction (SFE) is one of the most commonly used extraction techniques in the course of analysis or preparation. It is environmentally friendly and has some advantages over other conventional extraction methods. This review covers the recent developments of SFE in the extraction of essential oils from the plant materials during the period 2005 to 2011, in particular some factors influencing SFE extraction yield, its characteristics and applications. Key words: Supercritical fluid extraction, essential oils, review. INTRODUCTION Solvent extraction is one of the oldest methods of product contamination with solvent residues (Zaidul et al., separation known. The science of solvent extraction has 2006, 2007).SFE method is very advantageous and evolved over a long period of time and much progress environmentally friendly over other conventional either has been made in the understanding of solvation and the solvent or enzyme extraction methods for recovering properties of liquid mixtures used in extraction processes. -

Steam Distillation of Orange Oil
Chem 306 Name_______________________ Partners_______________________ _______________________ _______________________ Section (Circle) M Tu W Th Date_______________________ STEAM DISTILLATION OF ORANGE OIL Materials: 3 oranges, boiling chips, NaCl, ice, blender, grater, 50 mL buret, distillation apparatus, screw-top vial, ice, stirring rod, rinse bottle, vacuum grease, aluminum foil, cleaning brushes, detergent (Dawn) Purpose: In this laboratory activity you will separate orange oil from orange peel by the technique of steam distillation. Introduction: In this experiment we will be extracting D – limonene (4-Isopropeny-1- methyl-cyclohexene) from orange peel by use of steam distillation. Limonene belongs to a class of compounds called terpenes. Terpenes are a large and varied class of hydrocarbons, produced primarily by a wide variety of plants, particularly conifers. They are the major components of resin, and of turpentine produced from resin. The name "terpene" is derived from the word "turpentine". When terpenes are modified chemically, such as by oxidation or rearrangement of the carbon skeleton, the resulting compounds are generally referred to as terpenoids. Terpenes and terpenoids are the primary constituents of the essential oils of many types of plants and flowers. Essential oils are used widely as natural flavor additives for food, as fragrances in perfumery, in aromatherapy, and in traditional and alternative medicines. Synthetic variations and derivatives of natural terpenes and terpenoids also greatly expand the variety of aromas used in perfumery and flavors used in food additives. The building block of all terpenes is an isoprene unit. A limonene molecule contains two isoprene units. or “Isoprene” Units The terpene D-limonene is the major chemical component in orange oil and has a variety of uses. -

Steam Distillation of Toluene – Benzil Mixture
© Professor Kathleen V. Kilway and Robert Clevenger, Department of Chemistry, University of Missouri – Kansas City, 2006 2. Distillation J.R. Mohrig, Technique 12 Boiling points and distillation C.N. Hammond, 173-174 (general introduction) and P.F. Schatz: 176-180 (distillation and separation of mixtures) 194-196 (steam distillation) 203-205 (sources of confusion) Vocabulary: Vaporization The process of turning a liquid into vapor. Condensation The process of turning a vapor into a liquid. Azeotrope It is a mixture of two or more liquids, which boils at a constant boiling point. This procedure has been adapted from the macroscale procedure described in Macroscale and Microscale Organic Chemistry Experiments by Kenneth L. Williamson. In a nutshell: Normally, we distill solvents (the liquid in which we run the reaction) from a drying agent to dry the solvent if water is undesirable using a simple distillation. A fractional distillation is used to distill two or more components with similar boiling points. The Laboratory Techniques in Organic Chemistry’s authors (J.R. Mohrig, D.G. Alberg, G.E. Hofmeister, P.F. Schatz, and C.N. Hammond, pg 164) describes steam distillation as the “codistillation with water” and “allows the distillation of relatively nonvolatile organic compounds without complex vacuum systems; it is especially useful for separating volatile organic compounds from nonvolatile inorganic salts or from the leaves and seeds of plants”. Background What is distillation? It is the separation of liquids by vaporization and subsequent condensation of the vapors into a separate container. You are probably familiar with this process because it is used in the fragrance as well as the alcohol industry. -

Essential Oils and Steam Distillation
Plant Distillation Techniques Essential Oils and Steam distillation By Jill Mulvaney What is “Essential Oil” When I think about this, I sigh – and stare out the window at the birds on the tree, essential oils, such an enigma. Wikipedia describes them as “a concentrated hydrophobic liquid containing volatile aroma compounds from plants”, a pragmatic answer – a concentrated substance that is repelled from a mass of water (hydrophobic) that has smell or odour (aroma compounds). Apparently they are “essential” as well! When I’m out walking, I see and feel a plant, reach out and pick a leaf or flower, crush it in my palms, cup my hands, cover my mouth and nose and deeply inhale. The plant and I begin our conversation. We use the silent language of aroma and sensation. How I wish I could translate all that into academic words that make sense, but I have learnt to trust that the plant shares information with each aromatic conversation conducted with mindful body awareness and sensitivity. So maybe it is the soul part of the nature of plants that can speak. Aroma is certainly a powerful conduit of subtle change, 1. I have my Column Still response and reaction. Essential oils are aroma. set up in a sheltered spot in the To be able to steam distil plants I have found or grown myself has garden. I have already cleaned it been a profound experience. Even though the process is quite simple, by distilling rye flour slurry. To do you become part of a magical alchemy of transformation. The energy this I mix 450g of Rye Flour in 4 L of steam changes the chemistry of a plant so that its constituents are of water and pour into the pot. -

The Technique of Distillation
The Technique of Distillation Isolation of Volatile Oils from Spices by Steam Distillation and Bioassay for Biological Activity Introduction The spices cloves, cinnamon, ginger, nutmeg, and tumeric will be provided. There are many other possible spices such as caraway, cumin, or thyme. Feel free to bring in ~10 grams of a spice of your choice. There is no guarantee that a particular spice will yield much volatile oil or that the components of this oil will have biological activity, but it will be fun to see if they do. If you don’t get anything from your spice, you can continue the procedure with some clove oil that is available. Activities: Read the Introduction section of the Distillation Experiment found in Chapter 5 of the Lab Guide. In the first lab (Day One), you will use steam distillation to separate any volatile oils from your spice of choice; you may work in teams for this distillation only. In preparation for this, read the theory of steam distillation presented below. You will be using a variation of the distillation set-up as in Procedures 1A and 1B in Chapter 5 of the Lab Guide. In place of the liquid mixtures toluene/cyclohexane or ethanol/water, you will use water and your solid spice to carry out steam distillation. However, typical distillation data for simple and fractional distillation is provided below and you will plot these for your Final Report (see last page of this handout). You are responsible for understanding how simple and fractional distillations are done and the advantages and disadvantages of these types of distillations as well as steam distillation. -

Supercritical CO2 Extraction of Essential Oil from Kabosu
Suetsugu et al. Flavour 2013, 2:18 http://www.flavourjournal.com/content/2/1/18 RESEARCH Open Access Supercritical CO2 extraction of essential oil from Kabosu (Citrus sphaerocarpa Tanaka) peel Takuya Suetsugu1,3, Masahiro Tanaka2*, Hideo Iwai1,3, Teruaki Matsubara1,3, Yukihiro Kawamoto3,5, Chiho Saito4, Yoshito Sasaki4, Munehiro Hoshino2, Armando T Quitain1, Mitsuru Sasaki1, Junshi Sakamoto4 and Motonobu Goto5 Abstract Background: Citrus sphaerocarpa Hort. ex Tanaka is one of many popular sour citruses in Japan. Its juice processing peel residues contain a lot of useful compounds including essential oil. Our interests mainly focused on the extraction of this essential oil using supercritical carbon dioxide (SC-CO2), an environmentally benign and generally regarded as safe solvent that has many advantages such as low critical temperature, low viscosity, and easy separation from the extract. In this research, essential oil was extracted from Citrus sphaerocarpa Tanaka peel using SC-CO2 at extraction temperatures of 313 to 353 K and pressures of 10 to 30 MPa. Results: A maximum yield of 1.55% (by weight of wet sample) was obtained at the temperature of 353 K and the pressure of 20 MPa. The yield obtained by SC-CO2 method was over 13 times higher than that of the conventional cold-press method. Extracted essential oil was qualitatively analyzed using GC/MS, identifying 49 compounds including several non-polar and weakly polar hydrocarbons such as terpenoid, free fatty acid, and coumarin. Compared to the extracts obtained by the conventional methods, the extracts by SC-CO2 had lower content of monoterpenes and higher content of oxygenated compounds, sesquiterpenes, which strongly contribute to the aromatic characteristics of the extracts. -

Distillation • Distillation Is a Method of Separating Mixtures Based on Differences in Their Volatilities in a Boiling Liquid Mixture
Distillation • Distillation is a method of separating mixtures based on differences in their volatilities in a boiling liquid mixture. • Distillation is a unit operation, or a physical separation process, and not a chemical reaction. • Commercially, distillation is used to separate crude oil itinto more frac tions for spec ific uses suc h as transport, power generation and heating. • Water is distilled to remove impurities, such as salt from seawater. • Air is distilled to separate its components— oxygen, nitrogen, and argon—for industrial use. • Distillation of fermented solutions has been used since ancient times to produce distilled beverages with a higher alcohol content. History • The first clear evidence of distillation comes from GkGreek alhlchem itists working in Alexan dr ia ithfitin the first century AD. Distilled water has been known since at least ca. 200 AD. Arabs learned the process from the Egyptians and used it extensively in their alchemical experiments. • The first clear evidence from the distillation of alcohol comes from the School of Salerno in the 12th century. Fractional distillation was developed by Tadeo Alderotti in the 13th century. • In 1500, German alchemist Hieronymus Braunschweig published the Book of the Art of Distillation, the first book solely dedicated to the subject of distillation. In 1651, John French published The Art of Distillation the first major English compendium of practice. This includes diagrams with people in them showing the industrial rather than bench scale of the operation. • Early forms of distillation were batch processes using one vaporization and one condensation. Purity was improved by further distillation of the condensate. Greater volumes were processed by simply repeating the dis tilla tion. -
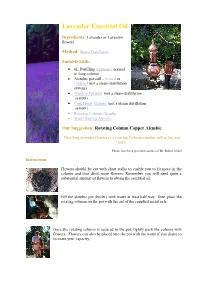
Using Your Rotating Column Copper Alembic Essential Oil Distiller
Lavender Essential Oil Ingredients: Lavender or Lavandin flowers Method: Steam Distillation Suitable Stills: • 6L Distilling Appliance normal or long column • Alembic pot still - riveted or soldered (not a steam distillation system) • Alquitar Pot Still (not a steam distillation system) • Cask Hood Alembic (not a steam distillation system) • Rotating Column Alembic • Water Sealing Alembic Our Suggestion: Rotating Column Copper Alembic Distilling lavender flowers in a rotating Column alembic still is fun and easy Photos have been provided courtesy of Mr. Robert Seidel Instructions: Flowers should be cut with short stalks to enable you to fit more in the column and thus distil more flowers. Remember you will need quite a substantial amount of flowers to obtain the essential oil. Fill the alembic pot (boiler) with water at least half way. Then place the rotating column on the pot with the aid of the supplied metal axle. Once the rotating column is secured to the pot, tightly pack the column with flowers. Flowers can also be placed into the pot with the water if you desire to increase your capacity. Flowers can also be packed tightly into the alembic lid. Simply turn over the lid, place the provided bowl-shaped sieve (not seen in the photo) at the bottom (the entrance to the piping section). Then pack the lid with flowers. The sieve will not let the flowers pass to the piping and thus clog the passage. Once the flowers have been packed in, assemble the lid on the column and connect the swan neck to the condensing recipient. In order to seal the still from steam leaks, a traditional effective sealing technique is used.