Acid-Base Behavior in 50-Percent Aqueous Methanol: Thermodynamics of the Dissociation of Protonated Tris(Hydroxymethyl)Aminomethane and Nature of the Solvent Effect*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Titration of Aspirin Tablets
Bellevue College | CHEM& 161 Titration of Aspirin Tablets In this lab, you will determine the percent purity of two commercially available aspiring tablets using an acid-base titration. In general, an acid and a base react to produce a salt and water by transferring a proton (H+): HA (aq) + NaOH (aq) H2O (l) + NaA (aq) (1) acid base salt The active ingredient in aspirin, and the chemical for which aspirin is the common name, is acetylsalicylic acid. To determine the amount of aspirin (acetylsalicylic acid) in a sample, the precise volume and concentration of the NaOH, and the overall reaction, must be known. The NaOH serves as a secondary standard, because its concentration can change over time. To find the precise concentration of the NaOH, it must be titrated against a primary standard, an acid that dissolves completely in water, has a high molar mass, that remains pure upon standing, and is not hygroscopic (tending to attract water from the air). Because sodium hydroxide is hygroscopic, it draws water from its surroundings. This mean one cannot simply weigh out a sample of sodium hydroxide, dissolve it in water, and determine the number of moles of sodium hydroxide present using the mass recorded, since any sample of sodium hydroxide is likely to be a mixture of sodium hydroxide and water. Thus, the most common way to determine the concentration of any sodium hydroxide solution is by titration. Determining the precise concentration of NaOH using a primary standard is called standardization. You will first standardize your NaOH solution, and then use it to analyze aspirin tablets for their aspirin content and purity. -

Chemistry 120: Experiment 1
Chemistry 120: Experiment 1 Preparation of a Standard Sodium Hydroxide Solution and Titration of Hydrochloric Acid In this experiment, we prepare solutions of NaOH and HCl which will be used in later experiments. We will require knowledge of the exact concentration of the two solutions, but it is not convenient either to weigh out solid NaOH or to measure out concentrated HCl. We do not know the composition of these materials well enough to obtain accurate numbers for our solutions by weighing them out. Instead, we take another approach. We will use the primary standard acid, potassium hydrogen phthalate, to make an acid solution containing a known amount of acid. The composition and purity of this acid is very well known, and we can accurately determine the amount of potassium hydrogen phthalate by weighing a sample. We can titrate this solid acid, after dissolving it in water, with our NaOH solution to accurately determine the NaOH concentration, as discussed below. This standardizes the NaOH solution against our primary standard. Then, with our standardized NaOH solution, we can titrate the HCl solution and accurately determine the HCl concentration. Thus, the concentrations determined for the NaOH and HCl solutions ultimately depend on the weight of our primary standard taken and the volumes of the solutions needed to react. Because the NaOH and HCl will be used to analyze other materials in future weeks, it is very important to determine their concentrations as carefully as possible. This suggests that careful weighing of the potassium hydrogen phthalate is very important. The presence of carbon dioxide is one reason why we cannot weigh out pure NaOH and use it as a primary standard. -

Year 12 ATAR Chemistry Unit 3: Acid Base Titrations
PROCESS TO EXCLUDE A STUDENT Year 12 ATAR Chemistry Unit 3: Acid Base Titrations Acids and Bases Except where indicated, this content © Department of Education Western Australia 2020 and released under Creative Commons CC BY NC Before re-purposing any third party content in this resource refer to the owner of that content for permission. https://creativecommons.org/licenses/by-nc/4.0/ Year 12 | ATAR | Chemistry | | Acid Base Titrations | © Department of Education WA 2020 Year 12 ATAR Chemistry Acids and Bases Chemistry ATAR Year 12 Unit 3 Acid Base Titrations Syllabus Points Covered This unit of work covers volumetric analysis used in acid-base titrations. Data obtained during titration can be used to calculate information about unknown solutions. Volumetric analysis methods involving acid-base reactions rely on the identification of an equivalence point by measuring the associated change in pH, using appropriate acid-base indicators or pH meters, to reveal an observable end point. Read the information below: Volumetric Analysis Acid-base reactions can be used to determine information about unknown solutions. An example could be the concentration of acetic (ethanoic) acid in a sample of vinegar or the acidity of a sample of biodiesel. In volumetric analysis, in order to work out the concentration of the acid in the solution, a chemist will react the acid with an alkaline solution. If the chemist knows the concentration of the alkali, and measures the volume of the alkali used to neutralise all of the acid, the chemist can calculate the number of moles of alkali used. Once this is known, the unknown number of moles of the acid can be worked out. -

REDOX TITRATIONS with IODINE Introduction
REDOX TITRATIONS WITH IODINE Introduction The shelf life of pharmaceutical products must always be tested. Hydrogen peroxide used in disinfectants is slowly reduced to water; it also photodegrades. Vitamin C in vitamin tablets is inherently unstable, being slowly oxidized by air. The ability to accurately and reliably determine the concentration of the active ingredients in various formulations is an important one. In this lab you will determine the content of hydrogen peroxide in a pharmaceutical disinfectant, as well as the concentration of ascorbic acid in Vitamin C supplemental tablets. Both analyses will involve the potentiometric titration of aqueous iodine with sodium thiosulfate using an automatic titrator. A platinum ring indicator electrode is used to follow the progress of the titration curve by potentiometry. Background Titrations Involving Iodine Iodine is a moderately weak oxidizing agent; it is reduced to form the iodide anion, as follows: – – I2(aq) + 2e l 2I (aq) E° = 0.621 V The above redox reaction is completely reversible, and so the iodide anion is a moderately weak reducing agent that will react with oxidizing analytes to produce iodine. Titrations involving iodine have evolved for the analysis of a number of oxidizing and reducing agents. In iodimetric titrations, the analyte (a reducing agent) reacts with iodine to produce iodide: – iodimetry Aox + I2 t Ared + 2I where Aox and Ared are the oxidized and reduced forms, respectively, of the analyte. In iodometric titrations, the analyte (an oxidizing agent) reacts with an unmeasured excess of iodide to produce iodine: – iodometry Ared + 2I t Aox + I2 excess The iodine produced in this reaction is stoichiometrically related to the amount of analyte originally present in the sample. -

Sodium Thiosulfate • This Method Is Called "Iodometry"
7/20/2010 Iodometric Determination of Copper In US One Cen t Co ins Ma de Be fore an d After 1982 XX YY Pennies are an interesting commodity. They are everywhere , an always present means of currency. The penny is ingrained in our society for many different social and economic reasons. 1 7/20/2010 Penny Composition • Pennies made from 1962 to 1982 consist of 95% copper and 5% zinc. These copper rich pennies are currently worth more as a metal source than their face value. • Pennies minted after 1982 to the present day are only made up of 2.5% copper. This copper is plated onto a zinc core that makes up 97.5% of the penny. • The determination of copper in both pennies made before and after 1982 will be determined using the same method. Introduction to Iodometric Determination 2 7/20/2010 Wet Chemical Analysis • MtdithittlMore accurate and precise than instrumental methods. • Systematic errors have been detected in the past and can be accounted. • More involved process • Help to learn new techniques • Reinforce the techniques learned so far Process selection - Iodine • UdidltUsed widely to quan tittiltitatively measure chemicals reactions • Many analytical procedures are based on the release or uptake • An analyte that is an oxidizing agent is added to excess iodide to produce iodine • The iodine produced is determined by titration with sodium thiosulfate • This method is called "iodometry" 3 7/20/2010 Iodometry • Chosen f or it s st rai ghtf orwar d proce dure • The reaction is rapid and quantitative • The starch used is a clear and determinant indicator • The reagents involved are all easily obtained and in good supply. -

20 Applications of Oxidation/Reduction Titrations 20A AUXILIARY OXIDIZING and REDUCING REAGENTS 20A-1 Auxiliary Reducing Reagents
20 Applications of Oxidation/Reduction Titrations 20A AUXILIARY OXIDIZING AND REDUCING REAGENTS 20A-1 Auxiliary Reducing Reagents Table 20-1 Uses of the Walden Reductor and the Jones Reductor Walden Jones Ag(s) + Cl- → AgCl(s) + e- Zn(Hg)(s) → Zn2+ + Hg + 2e- Fe3+ + e- → Fe2+ Fe3+ + e- →Fe2+ Cu2+ + e- → Cu+ Cu2+ + 2e- → Cu(s) + - + + - 3+ H2MoO4 + 2H + e → MoO2 + 2H2O H2MoO4 + 6H + 3e → Mo + 4H2O 2+ + - 4+ 2+ + - 4+ UO2 + 4H + 2e → U + 2H2O UO2 + 4H + 2e → U + 2H2O 2+ + - 3+ UO2 + 4H + 3e → U + 2H2O + + - 2+ + + - 2+ V(OH)4 + 2H + e → VO + 3H2O V(OH)4 + 4H + 3e → V + 4H2O 2+ 2+ + - 3+ TiO not reduced TiO + 2H + e → Ti + 4H2O Cr3+ not reduced Cr3+ + e- → Cr2+ 20A-2 Auxiliary Oxidizing Reagents - Sodium Bismuthate: Mn(II) → MnO4 + - + + NaBiO3(s) + 4H + 2e → BiO + Na + 2H2O Ammonium Peroxydisulfate, ammonium persulfate, (NH4)2S2O8 in acidic soln: Cr(III) → dichromate Ce(III) → Ce(IV) Mn(II) → permanganate 2- - 2- S2O8 + 2e → 2SO4 Eº = 2.01 V The oxidations are catalyzed by traces of silver ion. The excess reagent is readily decomposed by a brief period of boiling: 2- 2- + S2O8 + 2H2O → 4SO4 + O2 + 4H Sodium Peroxide and Hydrogen Peroxide + - H2O2 + 2H + 2e → 2H2O Eº = 1.78 V Fig. 20-1 boiling: H2O2 → 2H2O + 2O2(g) A Jones reductor. 20B APPLING STANDARD REDUCING AGENTS 20B-1 Iron (II) Solutions iron(II) ammonium sulfate, Fe(NH4)2(SO4)2.6H2O (Mohr's salt) iron(II) ethylenediamine sulfate, FeC2H4(NH3)2(SO4)2.4H2O (Oesper's salt) 137 Air-oxidation of iron (II) takes place rapidly in neutral solutions but is inhibited in the presence of acids, with the most stable preparations being about 0.5 M in H2SO4. -

Estimation of Cu(II) Using Sodium Thiosulphate Solution (Iodometrically) Bsc
Estimation of Cu(II) using sodium thiosulphate solution (Iodometrically) BSc. (H) Physics Semester-III GE: Chemistry Aim: Estimation of Cu(II) using sodium thiosulphate solution (iodometrically) Oxidation-Reduction (Redox) Reactions: ❖ Reaction involving change of oxidation number or transfer of electrons among the reacting substances. ❖ The standard solutions are either oxidising or reducing agents. ❖ The principal oxidising agents are KMnO4, K2Cr2O7, Ce(SO4)2, I2, KIO3, and KBrO3. ❖ Frequently used reducing agents are Fe(II) and Sn(II) compounds, Na2S2O3, As2O3, Hg2(NO3)2, VCl2 or VSO4, CrCl2 or CrSO4 & TiCl3 or Ti2(SO4)3. - + 2+ 2MnO4 + 5H2C2O4 + 6H 2Mn + 10CO2 + 8H2 (Pink) (Colourless) - 2+ + 2+ 3+ 2MnO4 + 10Fe + 16H 2Mn + 10Fe + 8H (Pink) (Colourless) 2+ + 3+ 3+ Cr2O72- + 6Fe + 14H 2Cr + 6Fe + 7H2O (Orange) (Green) CuSO4 + 4KI Cu2I2 + K2SO4 + (Blue) (White ppt) 2Na2S2O3 + I2 Na2S4O6 + 2NaI (Sod. Thiosulphate) (Sod. Tetrathionate) Iodine Titration ❖ The redox titration involving iodine directly or indirectly as an oxidizing agent are called iodine titrations. Iodimetric Titrations: Titrations with a standard solution of iodine. Applications of Iodimetric Titrations: ★ 2- Determination of S2O3 ★ 2- Determination of SO3 ★ 3- Determination of AsO3 ★ Determination of SnCl2 2Na2S2O3 + I2 Na2S4O6 + NaI Na2SO3 + I2 + H2O Na2SO4 + 2HI Na3AsO3 + I2 + H2O Na3AsO4 + 2HI Iodometric Titrations: Iodine titrations in which some oxidizing agent liberate I2 from an I- solution and then liberated I2 is titrated with a standard solution of reducing agent. Applications of Iodometric Titrations 3+ Estimation of CuSO4, K2Cr2O7, KMnO4, Fe , H2O2, Br2, Cl2 etc. CuSO4 + 4KI Cu2I2 + K2SO4 + I2 (Blue) (White ppt) 2Na2S2O3 + I2 Na2S4O6 + 2NaI (Sod. Thiosulphate) (Sod. Tetrathionate K2Cr2O7 + 4H2SO4 Cr2(SO4)3 + K2SO4 + 4H2O + 3O Orange Green 6KI + 3H2SO4 + 3O 3K2SO4 + 3H2O + 3I2 2Na2S2O3 + I2 Na2S4O6 + 2NaI DETECTION OF THE END POINT: ❖ In this titration a solution of starch is used as indicator. -
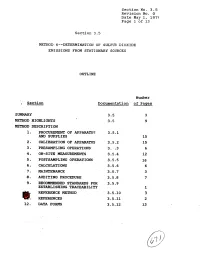
9. Recommended Standards for 3.5.9 Establishing Traceability 1 Ref'eresce Method 3.5-10 3 @!: Referexces 3.5.11 2 12
Section No. 3.5 Revision No. 0 Date May 1, 1975 Page 1 of 13 Section 3.5 METHOD. 6m-DETERMINATION OF SULFU?? DIOXIDE EMISSIONS FROM STATIONARY SOURCES OUTLINE Number ; Section Dscumenta.tion of Pages SUMMARY 3.5 3 METHOD HPGHLIGPFTS 3.5 9 METHOD DESCRIPTION 1. PRO- OF APPARATUS 3.5.1 AND SUPPLIES 15 2, CALIBRATION OF APP~TUS 3.5.2 15 3 - PMSAMPLING OPERATIONS 3. :.3 6 4. ON-SITE MEASUREMENTS 3.5.4 12 5. POSTSAMPLING OPERATIONS 3.5.5 16 6. CALCULATIONS 3.5.6 6 a. MAINTrnANCE 3.5-7 3 8. AUDITIN6 PROCEDURE 3.5.8 7 9. RECOMMENDED STANDARDS FOR 3.5.9 ESTABLISHING TRACEABILITY 1 REF'ERESCE METHOD 3.5-10 3 @!: REFEREXCES 3.5.11 2 12. DATA FORMS 3.5.12 13 Section No. 3-5 Revision No. 0 Date May 1, 1979 Page 2 of 13 SUMMARY This Method 6 test procedure is applicable to the determina- tion of sulfur dioxide emissions from stationary sources. A gas sample is extracted from the sampling point in the stack. The sulfur dioxide is separated from the sulfuric acid mist (includ- ing sulfur trioxide) and is measured by the barium-thorin titra- tion method. The barium ions react preferentially with sulfate ions in solution to form a highly insoluble barium sulfate pre- cipitate. When the barium has reacted with all sulfate ions, excess barium then reacts with the thorin indicator to form a metal salt of the indicator, resulting in a color change. The minimum detectable limit of the method has been de- termined to be 3.4 mg S02/m 3 (2.12 x lom7 lb S0,/ft3). -

VOLUMETRIC TITRATIONS I. Introduction A. Volumetric Methods
VOLUMETRIC TITRATIONS I. Introduction A. Volumetric methods are one of the three categories of titrimetric methods. B. Titrimetric methods are analytical procedures in which the amount of an analyte is determined from the amount of a standard reagent required to react completely with the analyte. C. The three types of titrimetric methods include: 1. Volumetric methods - measuring the volume of a solution (of known concentration) required to react completely with an analyte. 2. Gravimetric titrimetry - measuring the mass of a known reagent required to react completely with an analyte. 3. Coulometric titrimetry - measuring the electrical current (amps) produced when a known amount of reagent reacts completely with an analyte. II. Terminology of Volumetric Titrimetry. A. Standard solution (or standard titrant) - a reagent used to titrate the analyte. The reagent must: 1. Have a precisely known concentration. 2. Generally is added from a buret. B. Equivalent point - point in a titration where the added titrant is chemically equivalent to the analyte (i.e., reaction is at the stoichiometric ratio). C. End Point - a physical property (e.g., a color change) associated with the end point that it used to stop the titration. D. Titration error - the difference in added titrant volume between the end point and the equivalence point. Etitration = Vend point - Vequivalence point E. Indicator - a species added to the analyte to give an observable change (which is the end point) at or near the equivalence point. F. Back-titration - titrating the amount of excess titrant added using a second standard titrant. G. Primary standard - a highly purified compound that serves as a reference material. -

DETERMINATION of VITAMIN C in a PRODUCE PROTECTOR Iodometric Method - TEACHER
DETERMINATION OF VITAMIN C IN A PRODUCE PROTECTOR Iodometric method - TEACHER Since its discovery in the late 1920s1, no other chemical has ever been as celebrated as Vitamin C. The beneficial effect of Vitamin C is almost universally recognized2. It is a water- soluble keto-lactone with two ionizable hydroxyl groups; ascorbate monoanion (Figure - 1), AscH , is the dominant form at physiological pH and is is an excellent reducing agent as well. Research suggest that Vitamin C-rich foods, play an essential role against development of cancer; in addition, plasma concentrations of ascorbate have been shown inversely associated with cancer risk3,4 Figure 1 - ascorbate monoanion . Ascorbate monoanion undergoes two consecutive oxidations to form ascorbate radical (Asc ) and dehydroascorbic acid (DHA) as shown in Figure 2: Figure 2 - sequence of reactions which Vitamin C undergoes during its oxidation Fruit and vegetables are good sources of vitamin C, and ~90% of the daily intake in the general population comes from these sources. The content varies between species, but citrus fruit, kiwi, mango, and vegetables such as broccoli, tomatoes, and peppers are all rich sources of vitamin C. Since it degrades when heated and during storage, processing and preparation procedures should be considered when estimating dietary intake of vitamin C. In the small intestine, vitamin C reduces dietary iron and allows for efficient transport across the intestinal epithelium. In general, Vitamin C is safe and well tolerated, even in large doses. The U.S. Institute of medicine set the Tolerable Upper Intake Level for oral vitamin C ingestion at 2 g daily for adults. -
Preparation of Standard Sodium Hydroxide Solution and Titration of a Commercial Vinegar Product
Experiment 5 Chem 313 Preparation of Standard Sodium Hydroxide Solution and Titration of a Commercial Vinegar Product In this experiment we prepare a standard solution of NaOH, and then use it to determine the concentration of acetic acid in vinegar. We will require knowledge of the exact concentration of the NaOH solution, but it is not convenient either weigh solid NaOH, because it is hydroscopoic. Therefore, we must use a primary standard to determine the sodium hydroxide concentration to four significant figures. The primary standard that is most often used for standardizing sodium hydroxide solutions if potassium hydrogen phthalate, KHP. KHP of very high purity can be purchased, dried it in an oven, cooled it in a desiccator, and a sample can be accurately weighed on an analytical balance. After dissolving a known amount of KHP in distilled water, one can titrate the resulting solution with a sodium hydroxide solution. The volume of the sodium hydroxide solution required to reach the equivalence point and the mass of KHP is used to calculate the concentration of the sodium hydroxide solution to 4 or 5 significant figures. Once the NaOH solution is standardized it can be used to analyze acidic solutions, such as vinegar. The presence of carbon dioxide is another reason why NaOH can not be used as a primary standard. Water can readily absorb carbon dioxide. Dissolved carbon dioxide acts as a weak acid in aqueous solutions. CO2(g) ↔ CO2(aq) - + CO2(aq) + H2O H2CO3 HCO3 + H If basic solutions are prepared using water containing dissolved carbon dioxide, a portion - 2- of the base reacts with HCO3 to yield CO3 . -
Applications of Neutralization Titrations Applications of Neutralization Titrations
Chapter 16 Applications of Neutralization Titrations Applications of Neutralization Titrations Neutralization titrations are used to determine the concentration of acidic or basic analytes or analytes that can be converted to acids or bases by suitable treatment. Water is the usual solvent for neutralization titrations, because it is readily available, inexpensive, and nontoxic. Some analytes cannot be titrated in aqueous media because of low solubilities or because their strengths as acids or bases are not large enough to give satisfactory end points. Such substances can often be titrated by nonaqueous solvents, such as methyl and ethyl alcohol, glacial acetic acid, and methyl isobutyl ketone often make it possible to titrate such analytes in a solvent other than water. We shall restrict our discussions to aqueous systems. 16A Reagents for neutralization titrations Strong acids and strong bases produce the largest change in pH at the equivalence point. Hence, standard solutions for neutr. titrations are always prepared from these reagents 16A-1 Preparation of Standard Acid Solutions HCl solutions, Perchloric acid, and sulfuric acid are stable standard solutions. Restandardization is not required unless evaporation occurs. Standard solutions of nitric acid are seldom encountered because of their oxidizing properties To obtain most standard acid solutions, a solution of an approximate concentration is first prepared by diluting the concentrated reagent. The diluted acid solution is then standardized against a primary-standard base. A stock solution with a known HCl concentration can also be prepared by diluting a quantity of the concentrated reagent with an equal volume of water followed by distillation. 16A-2 The Standardization of Acids Acids are frequently standardized against weighed quantities of sodium carbonate.