HIV-1 Entry, Inhibitors, and Resistance
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Treatment-Drugs
© National HIV Curriculum PDF created September 23, 2021, 9:14 am Stavudine (Zerit) Table of Contents Stavudine Zerit Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Stavudine, an early nucleoside reverse transcriptase inhibitor (NRTI), was used as part of combination antiretroviral therapy for years, but now has become obsolete and replaced by better-tolerated and safer options. Stavudine poses risk of serious toxicity, including peripheral neuropathy (which can be permanent), pancreatitis, lipoatrophy, and lactic acidosis. Fatal and nonfatal cases of pancreatitis and lactic acidosis have been reported, especially when stavudine was combined with didanosine. According to the Adult and Adolescent ARV Guidelines, stavudine is no longer recommended for the treatment of HIV infection due to potential severe toxicity. Further, all persons currently taking stavudine should be strongly encouraged to switch to a safer medication. The sale and distribution of all strengths of stavudine will be discontinued and removed from the market in the United States in 2020. Key Clinical Trials Stavudine was studied for treatment-naïve patients as part of triple therapy, such as with lamivudine plus indinavir [START I], lamivudine plus lopinavir-ritonavir [M98-863], and lamivudine plus efavirenz [DART II]. Several studies demonstrated benefits of switching stavudine to newer NRTI agents, such as tenofovir disoproxil fumarate; the switch led to decreased rates of metabolic complications and mitochondrial toxicity [903E, SNAP, and ACTG 5142]. Resistance For a listing of the most common clinically significant mutations associated with stavudine (d4T) resistance, see the NRTI Resistance Notes on the Stanford University HIV Drug Resistance Database. Page 1/2 Key Drug Interactions For complete information on stavudine-related drug interactions, see the Drug Interactions section in the Stavudine (Zerit) Prescribing Information. -

Eparate Formulations According to the Prescribed Dosing Recommendations for These Products
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Lamivudine/Zidovudine Teva 150 mg/300 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each film-coated tablet contains 150 mg lamivudine and 300 mg zidovudine. For the full list of excipients see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet White, capsule shaped, biconvex, film-coated scored tablet – engraved with “L/Z” on one side and “150/300” on the other side. The tablet can be divided into equal halves. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Lamivudine/Zidovudine Teva is indicated in antiretroviral combination therapy for the treatment of Human Immunodeficiency Virus (HIV) infection (see section 4.2). 4.2 Posology and method of administration Therapy should be initiated by a physician experienced in the management of HIV infection. Lamivudine/Zidovudine Teva may be administered with or without food. To ensure administration of the entire dose, the tablet(s) should ideally be swallowed without crushing. For patients who are unable to swallow tablets, tablets may be crushed and added to a small amount of semi-solid food or liquid, all of which should be consumed immediately (see section 5.2). Adults and adolescents weighing at least 30 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one tablet twice daily. Children weighing between 21 kg and 30 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one-half tablet taken in the morning and one whole tablet taken in the evening. Children weighing from 14 kg to 21 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one-half tablet taken twice daily. -

HIV Tropism Testing for Maraviroc Response
Lab Management Guidelines V1.0.2021 HIV Tropism Testing for Maraviroc Response MOL.TS.185.A v1.0.2021 Introduction HIV tropism testing for maraviroc response is addressed by this guideline. Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline HIV-1 Tropism Phenotyping 87999 HIV-1 Tropism Genotyping, Common 87901 HIV-1 Tropism Genotyping, Other 87906 What is HIV tropism testing for Maraviroc response Definition HIV tropism testing is used to help determine an individual's response to maraviroc (Selzentry®). Maraviroc is only effective against CCR5-tropic HIV-1. Human immunodeficiency virus (HIV) HIV replicates itself in humans by infecting T-cells with CD4 receptors (often called CD4 cells). HIV-1 enters the CD4 cell by binding one of two cell surface co-receptors: CCR5 or CXCR4.1,2 Tropism classifications Tropism is the ability of HIV-1 virus to use one or both of these co-receptors. There are three main tropism classifications:3 o CCR5 tropism (R5-tropic) — HIV-1 virus that only infects cells with the CCR5 co-receptor. o CXCR4 tropism (X4-tropic) — HIV-1 virus that only infects cells with the CXCR4 co-receptor. © 2020 eviCore healthcare. All Rights Reserved. 1 of 5 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V1.0.2021 o Dual or mixed tropism (D/M-tropic) — HIV-1 virus populations that can use either co-receptor to infect cells. -
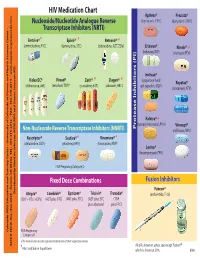
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

Review CCR5 Antagonists: Host-Targeted Antivirals for the Treatment of HIV Infection
Antiviral Chemistry & Chemotherapy 16:339–354 Review CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection Mike Westby* and Elna van der Ryst Pfizer Global R&D, Kent, UK *Corresponding author: Tel: +44 1304 649876; Fax: +44 1304 651819; E-mail: [email protected] The human chemokine receptors, CCR5 and suggest that these compounds have a long plasma CXCR4, are potential host targets for exogenous, half-life and/or prolonged CCR5 occupancy, which small-molecule antagonists for the inhibition of may explain the delay in viral rebound observed HIV-1 infection. HIV-1 strains can be categorised by following compound withdrawal in short-term co-receptor tropism – their ability to utilise CCR5 monotherapy studies. A switch from CCR5 to (CCR5-tropic), CXCR4 (CXCR4-tropic) or both (dual- CXCR4 tropism occurs spontaneously in approxi- tropic) as a co-receptor for entry into susceptible mately 50% of HIV-infected patients and has been cells. CCR5 may be the more suitable co-receptor associated with, but is not required for, disease target for small-molecule antagonists because a progression. The possibility of a co-receptor natural deletion in the CCR5 gene preventing its tropism switch occurring under selection pressure expression on the cell surface is not associated by CCR5 antagonists is discussed. The completion with any obvious phenotype, but can confer of ongoing Phase IIb/III studies of maraviroc, resistance to infection by CCR5-tropic strains – the aplaviroc and vicriviroc will provide further insight most frequently sexually-transmitted strains. into co-receptor tropism, HIV pathogenesis and The current leading CCR5 antagonists in clinical the suitability of CCR5 antagonists as a potent development include maraviroc (UK-427,857, new class of antivirals for the treatment of HIV Pfizer), aplaviroc (873140, GlaxoSmithKline) and infection. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

Reference ID: 2998411
• New onset or worsening renal impairment: Can include acute HIGHLIGHTS OF PRESCRIBING INFORMATION renal failure and Fanconi syndrome. Assess creatinine clearance These highlights do not include all the information needed to use (CrCl) before initiating treatment with COMPLERA. Monitor CrCl COMPLERA safely and effectively. See full prescribing and serum phosphorus in patients at risk. Avoid administering information for COMPLERA. COMPLERA with concurrent or recent use of nephrotoxic drugs. (5.3) COMPLERATM (emtricitabine/rilpivirine/tenofovir disoproxil • Caution should be given to prescribing COMPLERA with drugs fumarate) tablets that may reduce the exposure of rilpivirine. (5.4) Initial U.S. Approval: 2011 • Caution should be given to prescribing COMPLERA with drugs with a known risk of Torsade de Pointes. (5.4) WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT ACUTE • Depressive disorders: Severe depressive disorders (depressed EXACERBATION OF HEPATITIS B mood, depression, dysphoria, major depression, mood altered, negative thoughts, suicide attempt, suicidal ideation) have been See full prescribing information for complete boxed warning. reported. Immediate medical evaluation is recommended for severe depressive disorders. (5.5) • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of • Decreases in bone mineral density (BMD): Consider monitoring nucleoside analogs, including tenofovir disoproxil BMD in patients with a history of pathologic fracture -

Aplaviroc (APL, 873140)
Hepatotoxicity observed in clinical trials of aplaviroc (APL, 873140) WG Nichols1, HM Steel1, TM Bonny1, SS Min1, L Curtis1, K Kabeya2, N Clumeck2 1 GlaxoSmithKline, Research Triangle Park, USA; 2 CHU-Saint-Pierre, Brussels, Belgium. Aplaviroc (APL, 873140, Ono 4128) O CH3 O HO • Specific CCR5 antagonist O N OH N NH • Potent HIV entry inhibitor O – mean 1.66 log10 decline at nadir in HIV-RNA after 10d monotherapy1 • Safety profile supported further study in humans 1 Lalezari J et al. AIDS 2005;19:1443–1448. Aplaviroc Phase 2b Program CCR100136 CCR102881 • 195 treatment-naïve • 147 treatment-naïve subjects randomized to subjects randomized to – APL 200mg BID / LPV/r – APL 600mg BID / Combivir – APL 400mg BID / LPV/r – APL 800mg BID / Combivir – APL 800mg QD / LPV/r – Combivir / efavirenz – LPV/r / Combivir – 2:2:1 randomisation – 2:2:2:1 randomization Sentinel case: Severe hepatitis • 39 year old HIV+ male – CD4 283 cells/mm3 – HBV/HCV negative – Normal AST/ALT/bilirubin • APL 800mg BID + COM • Day 59: developed severe hepatic cytolysis • Liver biopsy: – Chronic inflammatory infiltrate, mod intensity Liver biopsy (portal area) – Consistent with drug-induced hepatotoxicity CCR102881 Individual Patient LFT Plots ALP ALT AST BILT CK 70 60 50 40 30 APL RX 20 LFT Values (x ULN) 10 0 -50 -40 -30 -20 -10 0 10 20 30 40 50 60 70 80 90 100 Study Day Review of Liver Enzyme Elevations in APL Phase IIb trials • 336 subjects received treatment – 282 subjects on APL – median duration of therapy: 13 wks • Central Lab database query to identify – any Grade -

Zero Dollar Cost Share – Generic Tenofovir Disoproxil Fumarate 300 Mg P&T Approval Date 11/2019, 8/2020, 9/2020 Effective Date 10/1/2020; Oxford Only: 11/1/2020
UnitedHealthcare Pharmacy Clinical Pharmacy Programs Program Number 2020 P 1289-3 Program Prior Authorization/Regulatory Medication HIV Pre-exposure Prophylaxis (PrEP) Zero Dollar Cost Share – generic tenofovir disoproxil fumarate 300 mg P&T Approval Date 11/2019, 8/2020, 9/2020 Effective Date 10/1/2020; Oxford only: 11/1/2020 . 1. Background: The U.S. Preventive Services Task Force (USPSTF) recommends that clinicians offer preexposure prophylaxis (PrEP) with effective antiretroviral therapy to persons who are at high risk of HIV acquisition.1 Once-daily oral treatment with Truvada® (emtricitabine/tenofovir disoproxil fumarate) is approved by the US Food and Drug Administration (FDA) for use as PrEP in persons at risk of sexual acquisition of HIV infection. Several studies reviewed by the USPSTF found that tenofovir disoproxil fumarate alone was also effective as PrEP and CDC guidelines note that, given these trial data, tenofovir disoproxil fumarate alone can be considered as an alternative regimen for high-risk heterosexually active men and women and persons who inject drugs.1-3 This program is designed to meet Health Care Reform requirements which require coverage of effective antiretroviral therapy, which includes tenofovir disoproxil fumarate or Truvada, at zero dollar cost share if being used for PrEP and criteria are met. 2. Coverage Criteria: A. Coverage at zero dollar cost share of generic tenofovir disoproxil fumarate 300 mg will be approved based on both of the following criteria: 1. Member is taking generic tenofovir disoproxil fumarate 300 mg as effective antiretroviral therapy for PrEP -AND- 2. Generic tenofovir disoproxil fumarate 300 mg will be used as part of a comprehensive prevention strategy including other prevention measures Authorization will be issued for zero copay with deductible bypass for 12 months. -

Design and Evaluation of 5•²-O-Dicarboxylic And
University of Rhode Island DigitalCommons@URI Open Access Master's Theses 2014 DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES Bhanu Priya Pemmaraju Venkata University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/theses Recommended Citation Pemmaraju Venkata, Bhanu Priya, "DESIGN AND EVALUATION OF 5′-O-DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES" (2014). Open Access Master's Theses. Paper 474. https://digitalcommons.uri.edu/theses/474 This Thesis is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Master's Theses by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. DESIGN AND EVALUATION OF 5′-O- DICARBOXYLIC AND POLYARGININE FATTY ACYL DERIVATIVES OF ANTI-HIV NUCLEOSIDES BY BHANU PRIYA, PEMMARAJU VENKATA A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE MASTER’S DEGREE IN BIOMEDICAL AND PHARMACEUTICAL SCIENCES UNIVERSITY OF RHODE ISLAND 2014 MASTER OF SCIENCE THESIS OF BHANU PRIYA, PEMMARAJU VENKATA APPROVED: Thesis Committee: Major Professor Keykavous Parang Roberta King Stephen Kogut Geoffrey D. Bothun Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT 2′,3′-Dideoxynucleoside (ddNs) analogs are the most widely used anti-HIV drugs in the market. Even though these drugs display very potent activities, they have a number of limitations when are used as therapeutic agents. The primary problem associated with ddNs is significant toxicity, such as neuropathy and bone marrow suppression.