New Zealand Data Sheet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effective Dose Range of Enalapril in Mild to Moderate Essential Hypertension
Br. J. clin. Pharmac. (1985), 19, 605-611 Effective dose range of enalapril in mild to moderate essential hypertension R. BERGSTRAND', H. HERLITZ2, SAGA JOHANSSON', G. BERGLUND2, A. VEDIN', C. WILHELMSSON', H. J. GOMEZ3, V. J. CIRILLO3 & J. A. BOLOGNESE4 'Department of Medicine, Ostra Hospital and 2Department of Medicine I, Sahlgrenska Hospital, Goteborg, Sweden and Department of 3Cardiovascular Clinical Research and 4Clinical Biostatistics, Merck Sharp & Dohme Research Laboratories, Rahway, New Jersey, USA 1 The dose-response relationship of enalapril was evaluated in a double-blind, balanced, two-period, incomplete-block study in 91 patients with mild to moderate essential hyper- tension. 2 Patients were randomly assigned to two of six treatments: placebo, 2.5, 5, 10, 20 and 40 mg/day of enalapril maleate. There were two 3-week treatment periods, each preceded by a 4-week, single-blind placebo washout. 3 Each dose of enalapril produced significant decreases in standing and supine systolic and diastolic blood pressure after 2 and 3 weeks of treatment. There were no significant changes on placebo. 4 There was a significant linear dose response relationship for both mean blood pressure and mean change from baseline in blood pressure (P < 0.01 for systolic and mean arterial pressure, and P < 0.05 for diastolic pressure). 5 Enalapril was associated with an increasing dose-response relationship across the 2.5- 40 mg/day range. The 2.5 mg/dose is effective in some patients; however, doses ¢ 10 mg/ day may be necessary to achieve satisfactory blood pressure control. Keywords enalapril angiotensin converting enzyme inhibitor dose-response relationship Introduction In recent years much interest has been focused with renal impairment treated with high doses of on angiotensin converting enzyme (ACE) in- captopril. -

The Direct Effects of the Angiotensin-Converting Enzyme
European Journal of Endocrinology (2006) 154 355–361 ISSN 0804-4643 EXPERIMENTAL STUDY The direct effects of the angiotensin-converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets Roberto Lupi, Silvia Del Guerra, Marco Bugliani, Ugo Boggi1, Franco Mosca1, Scilla Torri, Stefano Del Prato and Piero Marchetti Metabolic Unit, Department of Endocrinology and Metabolism, University of Pisa, Ospedale Cisanello, via Paradisa 2, 56100 Pisa, Italy and 1Transplant Unit, Department of Oncology, University of Pisa, Pisa, Italy, Menarini Ricerche SpA, Florence, Italy (Correspondence should be addressed to P Marchetti; Email: [email protected]) Abstract Objective: Data from prospective studies suggest a significant reduction in the risk of new diabetes from drug therapies containing angiotensin-converting enzyme (ACE) inhibitors. Since the renin–angio- tensin system (RAS) has been found locally in several tissues and cells, including pancreatic islets, we hypothesized that the positive metabolic effects of ACE inhibitors may be due to a beneficial action of these compounds on insulin-secreting b-cells. Design and methods: Isolated human pancreatic islets were studied after 24 h of incubation with 22.2 mmol/l glucose, with or without the presence in the incubation medium of 0.5–6.0 mmol/l zofe- noprilat or enalaprilat, ACE inhibitor drugs which differ by the presence of a sulphydryl or a carboxyl group in their structural formula. Functional and molecular studies were then performed to assess insulin secretion, redox balance, mRNA and protein expression. Results: Angiotensinogen, ACE and angiotensin type 1 receptor mRNA expression increased in islets cultured in high glucose; this was similarly prevented by the presence of either ACE inhibitor. -
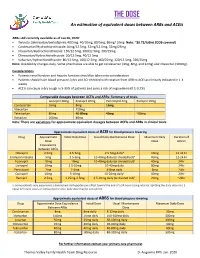
THE DOSE an Estimation of Equivalent Doses Between Arbs and Aceis
THE DOSE An estimation of equivalent doses between ARBs and ACEIs ARBs still currently available as of Jan 26, 2020: Twynsta (telmisartan/amlodipine): 40/5mg. 40/10mg, 80/5mg, 80mg/ 10mg Note: ~$0.73/tablet (ODB covered) Candesartan/Hydrochlorothiazide:16mg/12.5mg, 32mg/12.5mg, 32mg/25mg Irbesartan/Hydrochlorothiazide: 150/12.5mg, 300/12.5mg, 300/25mg Olmesartan/Hydrochlorothiaizde: 20/12.5mg, 40/12.5mg Valsartan/Hydrochlorothiazide: 80/12.5mg, 160/12.5mg, 160/25mg, 320/12.5mg, 320/25mg Note: Availability changes daily. Some pharmacies are able to get candesartan (4mg, 8mg, and 32mg) and irbesartan (300mg). Considerations Patients renal function and hepatic function should be taken into consideration Patients should have blood pressure, lytes and SCr checked with rotation from ARB to ACEI as clinically indicated in 1-4 weeks ACEIs can cause a dry cough in 5-35% of patients and carry a risk of angioedema (0.1-0.2%) Comparable dosages between ACEIs and ARBs- Summary of trials Lisinopril 20mg Enalapril 20mg Perindopril 4mg Ramipril 10mg Candesartan 16mg 8mg 16mg Irbesartan 150mg Telmisartan 80mg 40-80mg 40mg ~80mg Valsartan 160mg 80mg Note: There are variations for approximate equivalent dosages between ACEIs and ARBs in clinical trials. Approximate equivalent doses of ACEI for blood pressure lowering Drug Approximate Initial Daily Dose Usual Daily Maintenance Dose Maximum Daily Duration of Dose Dose Action Equivalence Between ACEIs Cilazapril 2.5mg 2.5-5mg 2.5-5mg dailya 10mg 12-24 hr Enalapril maleate 5mg 2.5-5mg 10-40mg daily (or divided bid)a 40mg 12-24 hr Fosinopril 10mg 10mg 10-40mg daily (or divided bid)a 40mg 24hr Lisinopril 10mg 2.5-10mg 10-40mg daily 80mg 24hr Perindopril 2mg 2-4mg 4-8mg daily 8mg 24hr Quinapril 10mg 5-10mg 10-20mg dailya 40mg 24hr Ramipril 2.5mg 1.25mg-2.5mg 2.5-10mg daily (or divided bid)a 20mg ~24hr a: Some patients may experience a diminished antihypertensive effect toward the end of a 24-hour dosing interval. -

Angiotensin-Converting Enzyme (ACE) Inhibitors
Angiotensin-Converting Enzyme (ACE) Inhibitors Summary Blood pressure reduction is similar for the ACE inhibitors class, with no clinically meaningful differences between agents. Side effects are infrequent with ACE inhibitors, and are usually mild in severity; the most commonly occurring include cough and hypotension. Captopril and lisinopril do not require hepatic conversion to active metabolites and may be preferred in patients with severe hepatic impairment. Captopril differs from other oral ACE inhibitors in its rapid onset and shorter duration of action, which requires it to be given 2-3 times per day; enalaprilat, an injectable ACE inhibitor also has a rapid onset and shorter duration of action. Pharmacology Angiotensin Converting Enzyme Inhibitors (ACE inhibitors) block the conversion of angiotensin I to angiotensin II through competitive inhibition of the angiotensin converting enzyme. Angiotensin is formed via the renin-angiotensin-aldosterone system (RAAS), an enzymatic cascade that leads to the proteolytic cleavage of angiotensin I by ACEs to angiotensin II. RAAS impacts cardiovascular, renal and adrenal functions via the regulation of systemic blood pressure and electrolyte and fluid balance. Reduction in plasma levels of angiotensin II, a potent vasoconstrictor and negative feedback mediator for renin activity, by ACE inhibitors leads to increased plasma renin activity and decreased blood pressure, vasopressin secretion, sympathetic activation and cell growth. Decreases in plasma angiotensin II levels also results in a reduction in aldosterone secretion, with a subsequent decrease in sodium and water retention.[51035][51036][50907][51037][24005] ACE is found in both the plasma and tissue, but the concentration appears to be greater in tissue (primarily vascular endothelial cells, but also present in other organs including the heart). -

Is Enalapril and Losartan Combination Irrational?Irrational?
Correspondence Is enalapril and losartan combination irrational?irrational? I read with great interest the editorial by C.S. Gautam and of exogenous angiotensin I pressor effects.[4] Therefore, the S. Aditya, which appeared in the Indian Journal of enalapril-losartan combinations are more potent at Pharmacology in June 2006 (vol, 38(3):167-70). A very achieving these goals than any of their constituents important topic was addressed in the editorial, and I individually. compliment the authors on this. On the other hand, I felt � Similarly synergistic efficacy of enalapril and losartan in confused when I found Enalapril + Losartan in the list of combination on exercise performance and oxygen irrational fixed dose combinations. I agree with the authors consumption at peak exercise in congestive heart failure that combining the two drugs affecting the same pathway is has been sugested.[5] irrational as it does not add to its efficacy, but I am not sure � The combination of an angiotensin-converting enzyme whether this combination should be categorised as irrational inhibitor and an angiotensin II receptor antagonist (AT1 or as rational. The results of ongoing research may answer receptor antagonist) in patients can significantly enhance this question in future. reduction in left ventricular hypertrophy. This can provide Apparently this combination may appear irrational, but greater protection to the heart against the overload caused actually rationality does exist for combining these two drugs. by persistent hypertension. Furthermore, the combination � From a physiological point of view, the general mechanism of these drugs did not increase the incidence of adverse of action of both angiotensin-converting enzyme inhibitor effects. -

Lisinopril (Ethics) Lisinopril Tablets 5 Mg, 10 Mg, 20 Mg
New Zealand Data Sheet Lisinopril (Ethics) Lisinopril Tablets 5 mg, 10 mg, 20 mg 1. NAME OF THE MEDICINAL PRODUCT LISINOPRIL (Ethics) 5 mg, tablet LISINOPRIL (Ethics) 10 mg, tablet LISINOPRIL (Ethics) 20 mg, tablet 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Lisinopril (Ethics) tablets come in three strengths and contain lisinopril dihydrate equivalent to 5 mg, 10 mg or 20 mg lisinopril. 3. PHARMACEUTICAL FORM 5 mg tablet: A light pink coloured, circular, biconvex uncoated tablet. The tablet has a break line and is embossed with ‘5’ on one side and embossed with ‘BL’ on the other side. 10 mg tablet: A light pink coloured, circular, biconvex uncoated tablet. The tablet is embossed with ‘10’ on one side and embossed with ‘BL’ on the other side. 20 mg tablet: A pink, circular, biconvex uncoated tablet. The tablet is embossed with ‘20’ on one side and embossed with ‘BL’ on the other side. Lisinopril dihydrate. The chemical name for lisinopril dihydrate is N-[N-[(1S)-1-carboxy-3- phenylpropyl]-L-lysyl]-L-proline dihydrate. Its structural formula is: C21H31N3O5.2H2O, Molecular weight: 441.53, CAS No.: 83915-83-7 Lisinopril dihydrate is a white to off-white crystalline powder that is soluble in water, sparingly soluble in methanol and practically insoluble in ethanol. A synthetic peptide derivative, lisinopril dihydrate is an oral long acting angiotensin converting enzyme inhibitor. It is a lysine analogue of enalaprilat (active metabolite of enalapril). 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Lisinopril is indicated in the treatment of essential hypertension and in renovascular hypertension. It may be used alone or concomitantly with other classes of antihypertensive agents. -
Enalaprilat Injection Has Been Used Concomitantly with Digitalis, Beta
EN-2436 EN-2436 Enalaprilat Injection, USP Enalaprilat Injection, USP Rx only Rx only USE IN PREGNANCY When used in pregnancy during the second and third trimesters, ACE inhibitors can cause injury and even death to the developing fetus. When pregnancy is detected, Enalaprilat Injection, USP should be discontinued as soon as possible. See WARNINGS, Fetal/Neonatal Morbidity and Mortality. DESCRIPTION Enalaprilat Injection, USP is a sterile aqueous solution for intravenous administration. Enalaprilat is an angiotensin converting enzyme inhibitor. It is chemically described as (S)-1-[N-(1-carboxy-3-phenylpropyl)-L-alanyl]-L-proline Hospira, Inc., Lake Forest, IL 60045 USA USA 60045 IL Forest, Lake Inc., Hospira, dihydrate. Its molecular formula is C18H24N2O5 • 2H2O and its structural formula is: Printed in USA USA in Printed Enalaprilat is a white to off-white, crystalline powder with a molecular weight of 384.43. It is sparingly soluble in methanol and slightly soluble in water. Each milliliter contains 1.25 mg enalaprilat (anhydrous equivalent); sodium chloride to adjust tonicity; benzyl alcohol, 9 mg, added as a preservative. May contain sodium hydroxide for pH adjustment. CLINICAL PHARMACOLOGY Enalaprilat, an angiotensin-converting enzyme (ACE) inhibitor when administered intravenously, is the active metabolite of the orally administered pro-drug, enalapril maleate. Enalaprilat is poorly absorbed orally. Mechanism of Action Intravenous enalaprilat, or oral enalapril, after hydrolysis to enalaprilat, inhibits ACE in human subjects and animals. ACE is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor substance, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex. Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. -

Switching Ace-Inhibitors
Switching Ace-inhibitors http://www.ksdl.kamsc.org.au/dtp/switching_ace_inhibitors.html Change to → Enalapril Quinapril Ramipril Change from ↓ (Once daily dosing) (Once daily dosing) (Once daily dosing) Captopril Captopril 12.5mg daily Enalapril 2.5mg1 Quinapril 2.5mg Ramipril 1.25mg Captopril 25mg daily Enalapril 5mg1 Quinapril 5mg Ramipril 1.25-2.5mg Captopril 50mg daily Enalapril 7.5mg1 Quinapril 10mg Ramipril 2.5-5mg Captopril 100mg daily Enalapril 20mg1 Quinapril 20mg Ramipril 5-10mg2 Captopril 150mg daily Enalapril 40mg Quinapril 40mg Ramipril 10mg Fosinopril Fosinopril 5mg daily Enalapril 5mg Quinapril 5mg Ramipril 1.25mg Fosinopril 10mg daily Enalapril 10mg Quinapril 10mg Ramipril 2.5mg Fosinopril 20mg daily Enalapril 20mg Quinapril 20mg Ramipril 5mg Fosinopril 40mg daily Enalapril 40mg Quinapril 40mg Ramipril 10mg Lisinopril Lisinopril 5mg daily Enalapril 5mg Quinapril 5mg Ramipril 1.25mg Lisinopril 10mg daily Enalapril 10mg Quinapril 10mg Ramipril 2.5mg Lisinopril 20mg daily Enalapril 20mg Quinapril 20mg Ramipril 5mg Lisinopril 40mg Enalapril 40mg Quinapril 40mg Ramipril 10mg Perindopril Perindopril 2mg daily Enalapril 5-10mg Quinapril 5-10mg Ramipril 2.5mg Perindopril 4mg daily Enalapril 10mg-20mg Quinapril 10mg-20mg Ramipril 5mg Perindopril 8mg daily Enalapril 20-40mg Quinapril 20-40mg Ramipril 10mg Trandolapril Trandolapril 0.5mg d Enalapril 5mg Quinapril 5mg Ramipril 1.25mg Trandolapril 1mg daily Enalapril 10mg Quinapril 10mg Ramipril 2.5mg Trandolapril 2mg daily Enalapril 20mg Quinapril 20mg Ramipril 5mg Trandolapril 4mg daily Enalapril 40mg Quinapril 40mg Ramipril 10mg There are few studies comparing equivalent doses of ACE-inhibitors, for specific indications. Therefore, the above recommendations are based on clinical experiences and are not specific for any indication. -

Valoración De La Terapia Combinada De Enalapril Con Irbesartán Sobre La Función Renal En Pacientes Diabéticos Tipo 2
Revista Médica de la Universidad de Costa Rica. Volumen 1, número 1, artículo 7, septiembre 2007. http://www.revistamedica.ucr.ac. Sección Estudiantil Valoración de la terapia combinada de Enalapril con Irbesartán sobre la función renal en pacientes diabéticos tipo 2 Rodríguez Hernández, Andrea.*, Rosenkrantz Amón, Shunit.*, Jiménez Loría, Federico**, Chaverri Fernández, José Miguel*** *Estudiantes de Farmacia. Universidad de Costa Rica, **Farmacéutico. Subdirector de Farmacia Hospital México, ***Farmacéutico. Centro Nacional de Información de Medicamentos. INIFAR. Facultad de Farmacia. Universidad de Costa Rica. Resumen: El presente trabajo pretende adicionar evidencia que respalde el uso concomitante de enalapril e irbersartán en los pacientes diabéticos del Hospital México. Métodos: Se seleccionó una muestra de pacientes diabéticos atendidos en el servicio de nefrología del Hospital México durante el periodo 2004-2006, valorándose en ellos, la función renal y la correlación existente con respecto al tratamiento recibido como nefroprotección, ya sea IECA o ARA II asociado a IECA. Resultados: La proporción de pacientes que utilizaron el tratamiento combinado y logran mejorar su función renal es mucho mayor que la proporción de pacientes que alcanzan esta mejoría dentro del grupo control o con monoterapia. Conclusiones: Los datos indican que existe una posible ventaja al utilizar el tratamiento combinado IECA – ARA II vs. la monoterapia con IECA como agentes renoprotectores. Palabras claves: Neuropatía diabética, ARA II, IECAS, diabetes, renoprotección. Recibido: Junio 2007. Aceptado: Septiembre. Publicado: Septiembre 2007. _____________________________________________________________________ 64 Revista electrónica publicada por la Escuela de Medicina de la Universidad de Costa Rica, 2060 San José, Costa Rica. ® All rights reserved. Revista Médica de la Universidad de Costa Rica. -

Therapeutic Class Overview Renin Inhibitors and Combinations
Therapeutic Class Overview Renin Inhibitors and Combinations INTRODUCTION Approximately 92.1 million American adults have at least one type of cardiovascular disease according to the 2017 American Heart Association Heart Disease and Stroke Statistics update. From 2004 to 2014, mortality associated with cardiovascular disease declined 25.3% (Benjamin et al, 2017). An estimated 85.7 million Americans or 34% of US adults aged ≥20 years have high blood pressure (BP). Hypertension is an independent risk factor for cardiovascular disease and increases the mortality risks of cardiovascular disease and other diseases (Benjamin et al, 2017). Lowering of BP has been shown to reduce the risk of fatal and nonfatal cardiovascular events including stroke and myocardial infarctions (MI) improving cardiovascular health and reducing cardiovascular risk also includes lipid control, diabetes management, smoking cessation, exercise, weight management, and limited sodium intake (Benjamin et al, 2017). Aliskiren (TEKTURNA®) is the only single entity direct renin inhibitor available in the United States (U.S.) and is Food and Drug Administration (FDA)-approved for the treatment of hypertension, either as monotherapy or in combination with other antihypertensive agents. Currently, only one combination renin inhibitor product is available in the US. This product combines the direct renin inhibitor, aliskiren, with a thiazide diuretic (TEKTURNA-HCT®) and is approved for hypertension. Studies have demonstrated that the combination of two inhibitors of the renin angiotensin system (RAS), including aliskiren, an angiotensin converting enzyme inhibitor (ACE-I) or an angiotensin II receptor blocker (ARB), provide no renal or cardiovascular benefits, and significant adverse events, particularly in patients with diabetes and/or renal insufficiency. -

Comparative Proteinuria Management of Different Angiotensin-Converting Enzyme Inhibitors Or Angiotensin Receptor Blockers for No
Comparative proteinuria management of different angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for normotensive patients with CKD: a Bayesian network meta-analysis Huizhen Ye*, Zhihao Huo*, Peiyi Ye, Guanqing Xiao, Zhe Zhang, Chao Xie and Yaozhong Kong Nephrology Department, The First People's Foshan Hospital, Foshan, Guangdong, China * These authors contributed equally to this work. ABSTRACT Background. Both angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) are blood pressure-lowering agents, but they are also being used to control proteinuria in early chronic kidney disease (CKD) patients. However, clinically, some patients present merely proteinuria without hypertension. No guidelines pointed out how to select treatments for proteinuria in normotensive patients. Thus, we conducted a Bayesian network analysis to evaluate the relative effects of different kinds of ACEI or ARB or their combination on proteinuria and blood pressure reduction. Methods. The protocol was registered in PROSPERO with ID CRD42017073721. A comprehensive literature database query was carried out systematically according to PICOS strategies. The primary outcome was reduction in proteinuria, and the secondary outcomes were eGFR reduction and blood pressure reduction. Random- effects pairwise and Bayesian network meta-analyses were used to estimate the effect of different regimens. Results. A total of 14 RCTs with 1,098 patients were included in the analysis. All Submitted 2 September 2019 treatment strategies of ACEI, ARB or their combination had significantly greater efficacy Accepted 16 January 2020 Published 12 March 2020 in reducing proteinuria than placebo in normotensive CKD patients. The combination therapy of olmesartan+temocapril had the highest probability (22%) of being the most Corresponding author Yaozhong Kong, [email protected] effective treatment to reduce proteinuria in normotensive CKD patients. -

Double-Blind Comparison of Eprosartan and Enalapril on Cough and Blood Pressure in Unselected Hypertensive Patients
Journal of Human Hypertension (1999) 13, 413–417 1999 Stockton Press. All rights reserved 0950-9240/99 $12.00 http://www.stockton-press.co.uk/jhh ORIGINAL ARTICLE Double-blind comparison of eprosartan and enalapril on cough and blood pressure in unselected hypertensive patients WJ Elliott, for the Eprosartan Study Group The effects of a new angiotensin receptor antagonist, with a 3.45-fold higher risk of definite cough (14/261 vs -Overall cough incidence (from spon .(0.018 ؍ eprosartan (200 or 300 mg b.i.d.) and enalapril (5–20 mg 4/259, P u.i.d.) on cough and blood pressure were compared in taneous reports from patients, or investigator’s a 26-week, double-blind, randomised, parallel-group, observation) was also more frequent with enalapril, as multicentre, international study involving 528 patients compared to eprosartan. Both agents reduced blood with hypertension. Uptitration of doses was based on pressure significantly compared to baseline, although clinic blood pressure measurements during the first 12 the eprosartan-treated group had a slightly higher weeks, after which hydrochlorothiazide (12.5–25 response rate (defined as sitting diastolic blood press- mg/day) could be added. The frequency and intensity of ure Ͻ90 mm Hg, or at least a 10 mm Hg reduction from cough was assessed by a standardised questionnaire baseline), both at end of titration (70.3% vs 62.6%, administered at each clinic visit. The primary end-point P Ͻ 0.05) and after 26 weeks (81.7% vs 73.5%, These data suggest that, in unselected .(0.018 ؍ was the incidence of persistent, dry cough not due to P upper respiratory infection; change in sitting diastolic hypertensive patients, eprosartan is associated with blood pressure and overall incidence of cough were less cough and a somewhat higher responder rate secondary end-points.