Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by Pparα
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SLC44A1 Transport of Choline and Ethanolamine in Disease
SLC44A1 Transport of Choline and Ethanolamine in Disease by Adrian Taylor A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Doctor of Philosophy in Human Health and Nutritional Sciences Guelph, Ontario, Canada © Adrian Taylor, April, 2019 ABSTRACT SLC44A1 TRANSPORT OF CHOLINE AND ETHANOLAMINE IN DISEASE Adrian Taylor Advisor(s): University of Guelph, 2019 Marica Bakovic Choline and ethanolamine are important molecules required for the de novo synthesis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) via the Kennedy pathway. Additionally, these two molecules are vital for maintaining both muscular and neurological function. The goal of this thesis was to gain insight into PC and PE metabolism with the use of unique metabolic disturbances ranging from obesity and genetic mutations in neurodegenerative disease. Firstly, the protective effects of choline supplementation on muscular function were investigated within the Pcyt2+/- mouse model. In Pcyt2+/- mice, substrate flow through the CDP-ethanolamine branch of the Kennedy pathway was diminished resulting in triacylglycerol (TAG) accumulation and obesity. Supplemental choline improved muscle function by altering the expression of genes devoted to reducing TAG synthesis and restoring energy homeostasis. With this new insight about the role of choline in regulating metabolism, the cellular uptake mechanism of choline was then analyzed. Skin fibroblasts from two patients with homozygous mutations in the SLC44A1 gene suffering from Neurodegeneration with Brain Iron Accumulation (NBIA) were utilized. In these fibroblasts, SLC44A1 expression and choline uptake were drastically diminished. Moreover, PC levels were unaffected while PE levels were diminished relative to control, an indication of perturbed phospholipid homeostasis. -

Chemicals in the Fourth Report and Updated Tables Pdf Icon[PDF
Chemicals in the Fourth National Report on Human Exposure to Environmental Chemicals: Updated Tables, March 2021 CDC’s Fourth National Report on Human Exposure to Environmental Chemicals: Updated Tables, March 2021 provides exposure data on the following chemicals or classes of chemicals. The Updated Tables contain cumulative data from national samples collected beginning in 1999–2000 and as recently as 2015-2016. Not all chemicals were measured in each national sample. The data tables are available at https://www.cdc.gov/exposurereport. An asterisk (*) indicates the chemical has been added since publication of the Fourth National Report on Human Exposure to Environmental Chemicals in 2009. Adducts of Hemoglobin Acrylamide Formaldehyde* Glycidamide Tobacco Alkaloids and Metabolites Anabasine* Anatabine* Cotinine Cotinine-n-oxide* Hydroxycotinine* Trans-3’-hydroxycotinine* 1-(3-Pyridyl)-1-butanol-4-carboxylic acid* Nicotine* Nicotine-N’-oxide* Nornicotine* Tobacco-Specific Nitrosamines (TSNAs) N’-Nitrosoanabasine (NAB)* N’-Nitrosoanatabine (NAT)* N’-Nitrosonornicotine (NNN)* Total 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol) (NNAL)* Volatile N-nitrosamines (VNAs) N-Nitrosodiethylamine (NDEA)* N-Nitrosoethylmethylamine (NMEA)* N-Nitrosomorpholine (NMOR)* N-Nitrosopiperidine (NPIP)* N-Nitrosopyrrolidine (NPYR)* Disinfection By-Products Bromodichloromethane Dibromochloromethane Tribromomethane (Bromoform) Trichloromethane (Chloroform) Personal Care and Consumer Product Chemicals and Metabolites Benzophenone-3 Bisphenol A Bisphenol F* Bisphenol -

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

Pfoa) Or Perfluorooctane Sulfonate (Pfos
National Toxicology Program NTP U.S. Department of Health and Human Services NTP Monograph Immunotoxicity Associated with Exposure to Perfluorooctanoic Acid or Perfluorooctane Sulfonate September 2016 NTP MONOGRAPH ON IMMUNOTOXICITY ASSOCIATED WITH EXPOSURE TO PERFLUOROOCTANOIC ACID (PFOA) OR PERFLUOROOCTANE SULFONATE (PFOS) September, 2016 Office of Health Assessment and Translation Division of the National Toxicology Program National Institute of Environmental Health Sciences National Institutes of Health U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Systematic Review of Immunotoxicity Associated with Exposure to PFOA or PFOS TABLE OF CONTENTS Table of Contents ...............................................................................................................................II List of Table and Figures ................................................................................................................... IV Abstract ............................................................................................................................................. 1 Peer Review Of The Draft NTP Monograph ......................................................................................... 2 Peer-Review Panel .............................................................................................................................. 2 Introduction ...................................................................................................................................... 3 Objective and Specific Aims ............................................................................................................... -

DNA Sequencing and Sorting: Identifying Genetic Variations
BioMath DNA Sequencing and Sorting: Identifying Genetic Variations Student Edition Funded by the National Science Foundation, Proposal No. ESI-06-28091 This material was prepared with the support of the National Science Foundation. However, any opinions, findings, conclusions, and/or recommendations herein are those of the authors and do not necessarily reflect the views of the NSF. At the time of publishing, all included URLs were checked and active. We make every effort to make sure all links stay active, but we cannot make any guaranties that they will remain so. If you find a URL that is inactive, please inform us at [email protected]. DIMACS Published by COMAP, Inc. in conjunction with DIMACS, Rutgers University. ©2015 COMAP, Inc. Printed in the U.S.A. COMAP, Inc. 175 Middlesex Turnpike, Suite 3B Bedford, MA 01730 www.comap.com ISBN: 1 933223 71 5 Front Cover Photograph: EPA GULF BREEZE LABORATORY, PATHO-BIOLOGY LAB. LINDA SHARP ASSISTANT This work is in the public domain in the United States because it is a work prepared by an officer or employee of the United States Government as part of that person’s official duties. DNA Sequencing and Sorting: Identifying Genetic Variations Overview Each of the cells in your body contains a copy of your genetic inheritance, your DNA which has been passed down to you, one half from your biological mother and one half from your biological father. This DNA determines physical features, like eye color and hair color, and can determine susceptibility to medical conditions like hypertension, heart disease, diabetes, and cancer. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

PFAS MCL Technical Support Document
ATTACHMENT 1 New Hampshire Department of Environmental Services Technical Background Report for the June 2019 Proposed Maximum Contaminant Levels (MCLs) and Ambient Groundwater Quality Standards (AGQSs) for Perfluorooctane sulfonic Acid (PFOS), Perfluorooctanoic Acid (PFOA), Perfluorononanoic Acid (PFNA), and Perfluorohexane sulfonic Acid (PFHxS) And Letter from Dr. Stephen M. Roberts, Ph.D. dated 6/25/2019 – Findings of Peer Review Conducted on Technical Background Report June 28, 2019 New Hampshire Department of Environmental Services Technical Background Report for the June 2019 Proposed Maximum Contaminant Levels (MCLs) and Ambient Groundwater Quality Standards (AGQSs) for Perfluorooctane sulfonic Acid (PFOS), Perfluorooctanoic Acid (PFOA), Perfluorononanoic Acid (PFNA), and Perfluorohexane sulfonic Acid (PFHxS) June 28, 2019 Table of Contents Abbreviations ................................................................................................................................................. i Acknowledgements ...................................................................................................................................... iii Section I. Executive Summary ....................................................................................................................... 1 Section II. Introduction ................................................................................................................................. 2 Section III. Reference Dose Derivation ........................................................................................................ -

Genome-Wide Analysis of Androgen Receptor Binding and Gene Regulation in Two CWR22-Derived Prostate Cancer Cell Lines
Endocrine-Related Cancer (2010) 17 857–873 Genome-wide analysis of androgen receptor binding and gene regulation in two CWR22-derived prostate cancer cell lines Honglin Chen1, Stephen J Libertini1,4, Michael George1, Satya Dandekar1, Clifford G Tepper 2, Bushra Al-Bataina1, Hsing-Jien Kung2,3, Paramita M Ghosh2,3 and Maria Mudryj1,4 1Department of Medical Microbiology and Immunology, University of California Davis, 3147 Tupper Hall, Davis, California 95616, USA 2Division of Basic Sciences, Department of Biochemistry and Molecular Medicine, Cancer Center and 3Department of Urology, University of California Davis, Sacramento, California 95817, USA 4Veterans Affairs Northern California Health Care System, Mather, California 95655, USA (Correspondence should be addressed to M Mudryj at Department of Medical Microbiology and Immunology, University of California, Davis; Email: [email protected]) Abstract Prostate carcinoma (CaP) is a heterogeneous multifocal disease where gene expression and regulation are altered not only with disease progression but also between metastatic lesions. The androgen receptor (AR) regulates the growth of metastatic CaPs; however, sensitivity to androgen ablation is short lived, yielding to emergence of castrate-resistant CaP (CRCaP). CRCaP prostate cancers continue to express the AR, a pivotal prostate regulator, but it is not known whether the AR targets similar or different genes in different castrate-resistant cells. In this study, we investigated AR binding and AR-dependent transcription in two related castrate-resistant cell lines derived from androgen-dependent CWR22-relapsed tumors: CWR22Rv1 (Rv1) and CWR-R1 (R1). Expression microarray analysis revealed that R1 and Rv1 cells had significantly different gene expression profiles individually and in response to androgen. -

Adipose Tissue NAPE-PLD Controls Fat Mass Development by Altering the Browning Process and Gut Microbiota
ARTICLE Received 11 Jul 2014 | Accepted 4 Feb 2015 | Published 11 Mar 2015 DOI: 10.1038/ncomms7495 OPEN Adipose tissue NAPE-PLD controls fat mass development by altering the browning process and gut microbiota Lucie Geurts1, Amandine Everard1,*, Matthias Van Hul1,*, Ahmed Essaghir2, Thibaut Duparc1, Se´bastien Matamoros1, Hubert Plovier1, Julien Castel3, Raphael G.P. Denis3, Marie Bergiers1, Ce´line Druart1, Mireille Alhouayek4, Nathalie M. Delzenne1, Giulio G. Muccioli4, Jean-Baptiste Demoulin2, Serge Luquet3 & Patrice D. Cani1 Obesity is a pandemic disease associated with many metabolic alterations and involves several organs and systems. The endocannabinoid system (ECS) appears to be a key regulator of energy homeostasis and metabolism. Here we show that specific deletion of the ECS synthesizing enzyme, NAPE-PLD, in adipocytes induces obesity, glucose intolerance, adipose tissue inflammation and altered lipid metabolism. We report that Napepld-deleted mice present an altered browning programme and are less responsive to cold-induced browning, highlighting the essential role of NAPE-PLD in regulating energy homeostasis and metabolism in the physiological state. Our results indicate that these alterations are mediated by a shift in gut microbiota composition that can partially transfer the phenotype to germ-free mice. Together, our findings uncover a role of adipose tissue NAPE-PLD on whole-body metabolism and provide support for targeting NAPE-PLD-derived bioactive lipids to treat obesity and related metabolic disorders. 1 Metabolism and Nutrition Research Group, WELBIO-Walloon Excellence in Life Sciences and BIOtechnology, Louvain Drug Research Institute, Universite´ catholique de Louvain, Avenue E. Mounier, 73 B1.73.11, 1200 Brussels, Belgium. 2 de Duve Institute, Universite´ catholique de Louvain, Avenue Hippocrate, 74 B1.74.05, 1200 Brussels, Belgium. -
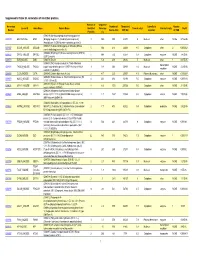
Supporting Table S3 for PDF Maker
Supplemental Table S3. Annotation of identified proteins. Number of Sequence Accession Number of Theoretical Subcellular Number Locus ID Gene Name Protein Name Identified Coverage Theoretical pI Protein Family NSAF Number Amino Acid MW (Da) Location of TMD Peptides (%) (O08539) Myc box-dependent-interacting protein 1 O08539 BIN1_MOUSE BIN1 (Bridging integrator 1) (Amphiphysin-like protein) 3 10.5 588 64470 5 Nucleus other NONE 5.73E-05 (Amphiphysin II) (SH3-domain-containing protein 9) (O08547) Vesicle-trafficking protein SEC22b (SEC22 O08547 SC22B_MOUSE SEC22B 5 30.4 214 24609 8.5 Cytoplasm other 2 0.000262 vesicle-trafficking protein-like 1) (O08553) Dihydropyrimidinase-related protein 2 (DRP-2) O08553 DPYL2_MOUSE DPYSL2 5 19.4 572 62278 6.4 Cytoplasm enzyme NONE 7.85E-05 (ULIP 2 protein) O08579 EMD_MOUSE EMD (O08579) Emerin 1 5.8 259 29436 5 Nucleus other 1 8.67E-05 (O08583) THO complex subunit 4 (Tho4) (RNA and transcription O08583 THOC4_MOUSE THOC4 export factor-binding protein 1) (REF1-I) (Ally of AML-1 1 9.8 254 26809 11.2 Nucleus NONE 2.21E-05 regulator and LEF-1) (Aly/REF) O08585 CLCA_MOUSE CLTA (O08585) Clathrin light chain A (Lca) 2 4.7 235 25557 4.5 Plasma Membrane other NONE 0.000287 (O08600) Endonuclease G, mitochondrial precursor (EC O08600 NUCG_MOUSE ENDOG 4 23.1 294 32191 9.5 Cytoplasm enzyme NONE 0.000134 3.1.30.-) (Endo G) (O08638) Myosin-11 (Myosin heavy chain, smooth O08638 MYH11_MOUSE MYH11 8 6.6 1972 227026 5.5 Cytoplasm other NONE 3.13E-05 muscle isoform) (SMMHC) (O08648) Mitogen-activated protein kinase kinase -

KONSTRUKTION VON ESCHERICHIA COLI PRODUKTIONSSTÄMMEN ZUR FERMENTATIVEN HERSTELLUNG VON SUCCINAT AUS GLYCERIN Stefan Söllner
KONSTRUKTION VON ESCHERICHIA COLI PRODUKTIONSSTÄMMEN ZUR FERMENTATIVEN HERSTELLUNG VON SUCCINAT AUS GLYCERIN Von der Fakultät 4 (Energie-, Verfahrens- und Biotechnik) der Universität Stuttgart zur Erlangung der Würde eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigte Abhandlung Vorgelegt von Stefan Söllner aus Schweinfurt Hauptberichter: Prof. Dr. rer. nat. Ralf Mattes Mitberichter: Prof. Dr.-Ing. Ralf Takors Tag der mündlichen Prüfung: 29.02.2012 Institut für Industrielle Genetik Universität Stuttgart 2012 Vielen Dank! … Herrn Prof. Dr. R. Mattes danke ich für die Überlassung des interessanten Themas, des Arbeitsplatzes, für gelegentliches Aufmuntern und für die Erstellung des Erstgutachtens dieser Arbeit. … Herrn Prof. Dr. R. Takors danke ich für die freundliche Übernahme des Zweitgutachtens und für spannende Diskussionen. … Herrn Dr. Josef Altenbuchner danke ich für die praktische Betreuung dieser Arbeit, für diverse Einladungen zu Grillfesten und ganz besonders für die intensive Durchsicht des Manuskriptes!!! … Herrn Dr. Martin Siemann-Herzberg danke ich für die motivierende, überschwängliche Begeisterung, die meine Ideen und Ergebnisse bei deren Besprechung jedesmal auslösten. … Herrn Prof. Dr. Reuss danke ich für die Initiierung des Projektes lange vor meiner Zeit. … Meinen Kollegen und Exkollegen danke ich für die gute Zusammenarbeit, das abwechslungsreiche Arbeitsklima sowie die vielen fachlichen und nichtfachlichen Gespräche, welche die Arbeit immer spannend gestalteten. Vor allem danke ich für das Verständnis für die von mir durchgeführten, absolut notwendigen, regelmäßigen Arbeitskontrollen. … Frau Dr. Anne Völker hat mir die Integration zu Beginn meines Aufenthaltes am IIG sehr erleichtert. Herzlichen Dank dafür! … Herrn Kambiz Morabbi Heravi danke ich recht herzlich für die Einladung ans National Institute of Genetic Engineering and Biotechnology in Teheran, Iran und für die internationale Freundschaft. -

Downloaded As a Text File, Is Completely Dynamic
BMC Bioinformatics BioMed Central Database Open Access ORENZA: a web resource for studying ORphan ENZyme activities Olivier Lespinet and Bernard Labedan* Address: Institut de Génétique et Microbiologie, CNRS UMR 8621, Université Paris-Sud, Bâtiment 400, 91405 Orsay Cedex, France Email: Olivier Lespinet - [email protected]; Bernard Labedan* - [email protected] * Corresponding author Published: 06 October 2006 Received: 25 July 2006 Accepted: 06 October 2006 BMC Bioinformatics 2006, 7:436 doi:10.1186/1471-2105-7-436 This article is available from: http://www.biomedcentral.com/1471-2105/7/436 © 2006 Lespinet and Labedan; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Despite the current availability of several hundreds of thousands of amino acid sequences, more than 36% of the enzyme activities (EC numbers) defined by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) are not associated with any amino acid sequence in major public databases. This wide gap separating knowledge of biochemical function and sequence information is found for nearly all classes of enzymes. Thus, there is an urgent need to explore these sequence-less EC numbers, in order to progressively close this gap. Description: We designed ORENZA, a PostgreSQL database of ORphan ENZyme Activities, to collate information about the EC numbers defined by the NC-IUBMB with specific emphasis on orphan enzyme activities.