Computational Functional Genomics-Based Ampliseq™ Panel for Next-Generation Sequencing of Key Genes of Pain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BIOINFORMATICS DOI: 10.1093/Bioinformatics/Btg1030
Vol. 19 Suppl. 1 2003, pages i222–i224 BIOINFORMATICS DOI: 10.1093/bioinformatics/btg1030 GeneLoc: exon-based integration of human genome maps Naomi Rosen, Vered Chalifa-Caspi, Orit Shmueli, Avital Adato, Michal Lapidot, Julie Stampnitzky, Marilyn Safran ∗ and Doron Lancet Weizmann Institute of Science, Rehovot, Israel Received on January 6, 2003; accepted on February 20, 2003 ABSTRACT to provide a comprehensive gene list, NCBI’s LocusLink Motivation: Despite the numerous available whole- contains thousands of model genes, categorized by level genome mapping resources, no comprehensive, inte- and type of support. Even known genes appearing in every grated map of the human genome yet exists. database may have different names in each database. The Results: GeneLoc, software adjunct to GeneCards and biologist must move among databases to figure out which UDB, integrates gene lists by comparing genomic coordi- genes are the same, and which could be a novel gene nates at the exon level and assigns unique and meaningful sought. UCSC’s Genome Browser website maps genes identifiers to each gene. from several sources on the same scale, but the maps are Availability: http://bioinfo.weizmann.ac.il/genecards and not integrated, making it difficult to relate genes from http://genecards.weizmann.ac.il/udb different sources. As stated (Jongeneel, 2000),‘there is an Supplementary information: http://bioinfo.weizmann.ac. urgent need for a human gene index that can be used to il/cards-bin/AboutGCids.cgi, http://genecards.weizmann. identify transcripts unambiguously.’ The author contends ac.il/GeneLocAlg.html that this index should have, among others, the following Contact: [email protected] qualities: comprehensiveness, uniqueness, and stability. -

Genome Analysis and Knowledge
Dahary et al. BMC Medical Genomics (2019) 12:200 https://doi.org/10.1186/s12920-019-0647-8 SOFTWARE Open Access Genome analysis and knowledge-driven variant interpretation with TGex Dvir Dahary1*, Yaron Golan1, Yaron Mazor1, Ofer Zelig1, Ruth Barshir2, Michal Twik2, Tsippi Iny Stein2, Guy Rosner3,4, Revital Kariv3,4, Fei Chen5, Qiang Zhang5, Yiping Shen5,6,7, Marilyn Safran2, Doron Lancet2* and Simon Fishilevich2* Abstract Background: The clinical genetics revolution ushers in great opportunities, accompanied by significant challenges. The fundamental mission in clinical genetics is to analyze genomes, and to identify the most relevant genetic variations underlying a patient’s phenotypes and symptoms. The adoption of Whole Genome Sequencing requires novel capacities for interpretation of non-coding variants. Results: We present TGex, the Translational Genomics expert, a novel genome variation analysis and interpretation platform, with remarkable exome analysis capacities and a pioneering approach of non-coding variants interpretation. TGex’s main strength is combining state-of-the-art variant filtering with knowledge-driven analysis made possible by VarElect, our highly effective gene-phenotype interpretation tool. VarElect leverages the widely used GeneCards knowledgebase, which integrates information from > 150 automatically-mined data sources. Access to such a comprehensive data compendium also facilitates TGex’s broad variant annotation, supporting evidence exploration, and decision making. TGex has an interactive, user-friendly, and easy adaptive interface, ACMG compliance, and an automated reporting system. Beyond comprehensive whole exome sequence capabilities, TGex encompasses innovative non-coding variants interpretation, towards the goal of maximal exploitation of whole genome sequence analyses in the clinical genetics practice. This is enabled by GeneCards’ recently developed GeneHancer, a novel integrative and fully annotated database of human enhancers and promoters. -

Transcriptional Regulation, Role in Inflammation and Potential Pharmacological Implications
UNIVERSITA’ DEGLI STUDI DI MILANO Dipartimento di Biotecnologie Mediche e Medicina Traslazionale Scuola di dottorato in Medicina Sperimentale e Biotecnologie Mediche Ciclo XXIX THE HUMAN-RESTRICTED DUPLICATED FORM OF THE α7 NICOTINIC ACETYLCHOLINE RECEPTOR, CHRFAM7A: TRANSCRIPTIONAL REGULATION, ROLE IN INFLAMMATION AND POTENTIAL PHARMACOLOGICAL IMPLICATIONS. Settore disciplinare: Bio/14 Tesi di dottorato di: Annalisa Maroli Relatore: Prof.ssa Grazia Pietrini Coordinatore: Prof. Massimo Locati Anno accademico: 2016-2017 1 2 Contents Abstract ................................................................................................................................. 5 1. Introduction ................................................................................................................... 7 1.1. Inflammation........................................................................................................... 7 1.1.1. Acute inflammation ......................................................................................... 7 1.1.2. Molecular mechanisms of acute inflammation ............................................... 9 1.1.3. Resolution of acute inflammation ................................................................. 13 1.1.4. Chronic inflammation .................................................................................... 14 1.2. The Cholinergic Anti-Inflammatory Pathway........................................................ 14 1.3. The α7 nicotinic acetylcholine receptor .............................................................. -

Full Review Molecular Disease Presentation in Diabetic Nephropathy
Nephrol Dial Transplant (2015) 30: iv17–iv25 doi: 10.1093/ndt/gfv267 Full Review Molecular disease presentation in diabetic nephropathy Downloaded from https://academic.oup.com/ndt/article/30/suppl_4/iv17/2324968 by guest on 25 September 2021 Andreas Heinzel1, Irmgard Mühlberger1, Gil Stelzer2, Doron Lancet2, Rainer Oberbauer3, Maria Martin4 and Paul Perco1 1emergentec biodevelopment GmbH, Vienna, Austria, 2Weizmann Institute of Science, Rehovot, Israel, 3Medical University of Vienna, Vienna, Austria and 4EMBL-EBI, Wellcome Trust Genome Campus, Hinxton, UK Correspondence and offprint requests to: Paul Perco; E-mail: [email protected] ABSTRACT INTRODUCTION Diabetic nephropathy, as the most prevalent chronic disease of Kidney disease is a common problem in patients with diabetes. the kidney, has also become the primary cause of end-stage Depending on the degree of GFR impairment and/or albumin- renal disease with the incidence of kidney disease in type 2 dia- uria, roughly 30% of subjects are affected. As with most betics continuously rising. As with most chronic diseases, the diabetes-associated comorbidities, the pathophysiology is pathophysiology is multifactorial with a number of deregulated multifactorial and the molecular pathways involved in the ini- molecular processes contributing to disease manifestation and tiation and progression constitute a wide and complex as well as progression. Current therapy mainly involves interfering in the redundant network of regulators. Considerable success has renin–angiotensin–aldosterone -

Causal Varian Discovery in Familial Congenital Heart Disease - an Integrative -Omic Approach Wendy Demos Marquette University
Marquette University e-Publications@Marquette Master's Theses (2009 -) Dissertations, Theses, and Professional Projects Causal Varian discovery in Familial Congenital Heart Disease - An Integrative -Omic Approach Wendy Demos Marquette University Recommended Citation Demos, Wendy, "Causal Varian discovery in Familial Congenital Heart Disease - An Integrative -Omic Approach" (2012). Master's Theses (2009 -). 140. https://epublications.marquette.edu/theses_open/140 CAUSAL VARIANT DISCOVERY IN FAMILIAL CONGENITAL HEART DISEASE – AN INTEGRATIVE –OMIC APPROACH by Wendy M. Demos A Thesis submitted to the Faculty of the Graduate School, Marquette University, in Partial Fulfillment of the Requirements for the Degree of Master of Science Milwaukee, Wisconsin May 2012 ABSTRACT CAUSAL VARIANT DISCOVERY IN FAMILIAL CONGENITAL HEART DISEASE – AN INTEGRATIVE –OMIC APPROACH Wendy M. Demos Marquette University, 2012 Background : Hypoplastic left heart syndrome (HLHS) is a congenital heart defect that leads to neonatal death or compromised quality of life for those affected and their families. This syndrome requires extensive medical intervention for the affected to survive. It is characterized by significant underdevelopment or non-existence of the components of the left heart and the aorta, including the left ventricular cavity and mass. There are many factors ranging from genetics to environmental relationships hypothesized to lead to the development of the syndrome, including recent studies suggesting a link between hearing impairment and congenital heart defects (CHD). Although broadly characterized those factors remain poorly understood. The goal of this project is to systematically utilize bioinformatics tools to determine the relationships of novel mutations found in exome sequencing to a familial congenital heart defect. Methods A systematic genomic and proteomic approach involving exome sequencing, pathway analysis, and protein modeling was implemented to examine exome sequencing data of a patient with HLHS. -

A New Cerkl Mouse Model Generated by CRISPR-Cas9 Shows
Retina ANewCerkl Mouse Model Generated by CRISPR-Cas9 Shows Progressive Retinal Degeneration and Altered Morphological and Electrophysiological Phenotype Elena B. Domènech,1,2 Rosa Andrés,1,2 M. José López-Iniesta,1 Serena Mirra,1,2 Rocío García-Arroyo,1 Santiago Milla,3 Florentina Sava,1 Jordi Andilla,4 Pablo Loza-Álvarez,4 Pedro de la Villa,3,5 Roser Gonzàlez-Duarte,1,2,6 and Gemma Marfany1,2,6,7 1Department of Genetics, Microbiology and Statistics, University of Barcelona, Barcelona, Spain 2CIBERER/ISCIII, University of Barcelona, Barcelona, Spain 3Department of Systems Biology, University of Alcalá, Madrid, Spain 4ICFO–The Institute of Photonic Sciences, Barcelona Institute of Science and Technology, Barcelona, Spain 5Ramón y Cajal Institute for Health Research, Madrid, Spain 6DBGen Ocular Genomics, Barcelona, Spain 7Institute of Biomedicine, University of Barcelona, Barcelona, Spain Correspondence: Gemma Marfany, PURPOSE. Close to 100 genes cause retinitis pigmentosa, a Mendelian rare disease that Avinguda Diagonal, 643, edifici affects 1 out of 4000 people worldwide. Mutations in the ceramide kinase-like gene Prevosti, planta 1, 08028 Barcelona, (CERKL) are a prevalent cause of autosomal recessive cause retinitis pigmentosa and Spain; cone–rod dystrophy, but the functional role of this gene in the retina has yet to be fully [email protected]. determined. We aimed to generate a mouse model that resembles the phenotypic traits of Received: January 20, 2020 patients carrying CERKL mutations to undertake functional studies and assay therapeutic Accepted: June 14, 2020 approaches. Published: July 13, 2020 METHODS. The Cerkl locus has been deleted (around 97 kb of genomic DNA) by gene Citation: Domènech EB, Andrés R, editing using the CRISPR-Cas9 D10A nickase. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

MTHFR) and Methionine Synthase Reductase (MTRR
ANTICANCER RESEARCH 28 : 2807-2812 (2008) Methylenetetrahy drofolate Reductase (MTHFR) and Methionine Synthase Reductase (MTRR) Gene Polymorphisms as Risk Factors for Hepatocellular Carcinoma in a Korean Population SUN YOUNG KWAK 1,2 , UN KYUNG KIM 3, HYO JIN CHO 2, HEE KEUN LEE 3, HYE JIN KIM 2, NAM KEUN KIM 2 and SEONG GYU HWANG 1,2 1Department of Internal Medicine, 2Institute for Clinical Research, College of Medicine, Pochon CHA University, Seongnam; 3Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, South Korea Abstract. Hepatocellular carcinoma (HCC) is the third were increased risk factors for the disease. The MTHFR most frequent cause of cancer death in South Korea, but 1298A>C and the MTRR 66A>G genotypes were associated genetic susceptibility factors of HCC have not been examined with an increased risk of HCC in th is Korean population. extensively. Methylenetetrahydrofolate reductase (MTHFR) Further studies involving larger and varied populations and methionine synthase reductase (MTRR) play an essential could provide a potential tool for cancer risk assessment in role in both DNA synthesis and methylation and patients who are at risk of developing HCC. polymorphisms in the MTHFR gene, 677C>T, 1298A>C and the MTRR gene, 66A>G, are associated with several types of Hepatocellular carcinoma (HCC) is one of the most common malignancy. In this study, the allelic frequencies and malignant tumors with the third highest mortality rate among genotype distribution of three polymorphisms in the MTHFR malignancies in South Korea. According to a statistical report and MTRR genes from 96 hepatocellular carcinoma (HCC) in 2006, it occurred in 33.8/10,000 men and 11.2/10,000 patients and 201 controls were examined to assess the women in South Korea in 2005 (1). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
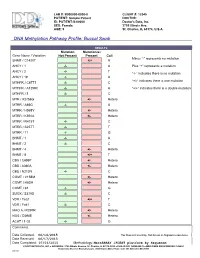
DNA Methylation Pathway Profile; Buccal Swab
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-00000 Doctor's Data, Inc. SEX: Female 3755 Illinois Ave. AGE: 5 St. Charles, IL 60174, U.S.A. !DNA Methylation Pathway Profile; Buccal Swab RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call Minus “-“ represents no mutation SHMT / C1420T +/+ A AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A “+/-“ indicates there is one mutation MTHFR / C677T -/- C MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G -/- A MTRR / H595Y +/- Hetero MTRR / K350A +/- Hetero MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 -/- G BHMT / 1 -/- A BHMT / 2 -/- C BHMT / 4 +/- Hetero BHMT / 8 +/+ T CBS / C699T +/- Hetero CBS / A360A +/- Hetero CBS / N212N -/- C COMT / V158M +/- Hetero COMT / H62H +/- Hetero COMT / 61 -/- G SUOX / S370S -/- C VDR / Taq1 +/+ T VDR / Fok1 -/- C MAO A / R297R +/- Hetero NOS / D298E +/- Hetero ACAT / 1-02 -/- G Comments: Date Collected: 06/14/2015 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 06/17/2015 Date Completed: 07/03/2015 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol .