A Case Report of a Girl with Polyarticular Disease Refractory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Mutations and Distribution of Single Nucleotide Polymorphic Alleles in the Human -L-Iduronidase (IDUA) Gene
article November/December 2002 ⅐ Vol. 4 ⅐ No. 6 Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human ␣-L-iduronidase (IDUA) gene Peining Li, PhD1,3, Tim Wood, PhD1,4, and Jerry N. Thompson, PhD1,2 Purpose: Mucopolysaccharidosis type I (MPS I) is an autosomal recessive disorder resulting from a deficiency of the lysosomal glycosidase, ␣-L-iduronidase (IDUA). Patients with MPS I present with variable clinical manifestations ranging from severe to mild. To facilitate studies of phenotype-genotype correlation, the authors performed molecular studies to detect mutations in MPS I patients and characterize single nucleotide polymorphism (SNP) in the IDUA gene. Methods: Twenty-two unrelated MPS I patients were subjects for mutation detection using reverse transcriptional polymerase chain reaction (RT-PCR) and genomic PCR sequencing. Polymorphism analyses were performed on controls by restriction enzyme assays of PCR amplicons flanking nine IDUA intragenic single nucleotide polymorphic alleles. Results: Eleven different mutations including two common mutations (Q70X, W402X), five recurrent mutations (D315Y, P533R, R621X, R628X, S633L), and four novel mutations (R162I, G208D, 1352delG, 1952del25bp) were identified from MPS I patients. Multiple SNP alleles coexisting with the disease-causing mutations were detected. Allelic frequencies for nine SNP alleles including A8, A20, Q33H, L118, N181, A314, A361T, T388, and T410 were determined. Conclusions: The results provide further evidence for the mutational heterogeneity among MPS I patients and point out possible common haplotype structures in the IDUA gene. Genet Med 2002:4(6):420–426. Key Words: mucopolysaccharidosis type I, ␣-L-iduronidase (IDUA) gene, mutations, single nucleotide polymor- phism, haplotype Mucopolysaccharidosis type I (MPS I, MIM 252800) is an and results in lysosomal accumulation and excessive urinary autosomal recessive disorder caused by various lesions in the excretion of partially degraded dermatan sulfate and heparan ␣-L-iduronidase (IDUA) gene. -

Mucopolysaccharidosis Type I
diagnostics Review Mucopolysaccharidosis Type I Francyne Kubaski 1,2,3,4 , Fabiano de Oliveira Poswar 1,2 , Kristiane Michelin-Tirelli 2,4 , Ursula da Silveira Matte 1,3,4,5,6, Dafne D. Horovitz 7, Anneliese Lopes Barth 7, Guilherme Baldo 1,3,4,5,8, Filippo Vairo 9,10 and Roberto Giugliani 1,2,3,4,5,6,11,* 1 Postgraduate Program in Genetics and Molecular Biology, UFRGS, Porto Alegre 91501970, Brazil; [email protected] (F.K.); [email protected] (F.d.O.P.); [email protected] (U.d.S.M.); [email protected] (G.B.) 2 Medical Genetics Service, HCPA, Porto Alegre 90035903, Brazil; [email protected] 3 INAGEMP, Porto Alegre 90035903, Brazil 4 Biodiscovery Research Group, Experimental Research Center, HCPA, Porto Alegre 90035903, Brazil 5 Gene Therapy Center, HCPA, Porto Alegre 90035903, Brazil 6 Department of Genetics, UFRGS, Porto Alegre 91501970, Brazil 7 Medical Genetics Department, National Institute of Women, Children, and Adolescent Health, Oswaldo Cruz Foundation, Rio de Janeiro 21040900, Brazil; [email protected] (D.D.H.); [email protected] (A.L.B.) 8 Department of Physiology, UFRGS, Porto Alegre 90050170, Brazil 9 Center for Individualized Medicine, Mayo Clinic, Rochester, MN 55905, USA; [email protected] 10 Department of Clinical Genomics, Mayo Clinic, Rochester, MN 55905, USA 11 Postgraduation Program in Medicine, Clinical Sciences, UFRGS, Porto Alegre 90035003, Brazil * Correspondence: [email protected]; Tel.: +55-51-3359-6338; Fax: +55-51-3359-8010 Received: 31 January 2020; Accepted: 10 March 2020; Published: 16 March 2020 Abstract: Mucopolysaccharidosis type I (MPS I) is caused by the deficiency of α-l-iduronidase, leading to the storage of dermatan and heparan sulfate. -

MPS1) (Hurler / Scheie Syndrome
Mucopolysaccharidosis 1 (MPS1) (Hurler / Scheie syndrome) Contact details Introduction Molecular Genetics Service MPS1 (MIM 252800) is an autosomal recessive lysosomal storage disorder, also Level 6, Barclay House known as Hurler syndrome (severe) or Scheie syndrome (milder variant). The 37 Queen Square condition is caused by a deficiency of the enzyme alpha-L-iduronidase (IDUA), which London, WC1N 3BH is required for lysosomal degradation of the glycosaminoglycans, heparin sulphate T +44 (0) 20 7762 6888 and dermatan sulphate. Affected individuals have a characteristic pattern of urine F +44 (0) 20 7813 8578 metabolites and a deficiency in the IDUA enzyme activity; biochemical enzyme analysis utilises these features to confirm a clinical diagnosis. Samples required Hurler patients are usually diagnosed by the age of 2 years and characteristically 5ml venous blood in plastic EDTA have short stature, coarse facial features, developmental delay, heart defects and bottles (>1ml from neonates) hepatosplenomegaly. Scheie patients can present at a later age, and have a milder Prenatal testing must be arranged course of symptoms, including joint stiffness, corneal clouding and aortic valve in advance, through a Clinical disease. Other patients have an intermediate phenotype. The phenotypic Genetics department if possible. heterogeneity correlates to some extent with the different nature of the mutations identified in the IDUA gene, although many novel mutations are known, there are Amniotic fluid or CV samples should be sent to Cytogenetics for ‘common’ mutations within the gene. dissecting and culturing, with The IDUA gene (4p16.3) has 14 exons and mutations have been found throughout instructions to forward the sample to the Regional Molecular Genetics the gene. -

Illinois Department of Public Health
NEWBORN SCREENING OFFICE OF HEALTH PROMOTION 535 W. Jefferson St., 2nd Floor Springfield, IL 62761 Phone: 217-785-8101 Fax: 217-557-5396 Mucopolysaccharidosis Type I (MPS I) Disease (Hurler, Hurler-Scheie and Scheie Syndromes) Information for Physicians and Other Health Care Professionals Definition MPS I disease, also frequently referred to as Hurler syndrome, is an inherited, autosomal recessive lysosomal storage disorder caused by deficiency in the activity of the enzyme alpha-L-iduronidase. This enzyme is responsible for the breakdown of certain glycosaminoglycans (GAGs). Lysosomal accumulation of these GAG molecules results in cell, tissue and organ dysfunction. Clinical Symptoms MPS I is a multisystem disorder and presents in three types with a wide range of symptoms. The severe form, MPS I H, also known as Hurler syndrome, has more severe symptoms that usually start within the first year of life. Symptoms of MPS I may include mental retardation and developmental delays, short stature, stiff joints, speech and hearing impairment, heart and lung disease, enlarged liver and spleen, hernia, coarse facial features, hydrocephalus, spinal compression, pain and a shortened life span. The other subtypes of MPS I are MPS I H-S (Hurler-Scheie syndrome) and MPS I S (Scheie syndrome). Children with MPS I H-S and MPS I S may have normal intelligence with milder symptoms starting later in childhood. Newborn Screening and Definitive Diagnosis In Illinois, newborn screening for MPS I disease is performed by measuring alpha-L-iduronidase enzyme activity. If newborn screening results indicate abnormal activity of this enzyme, referral should be made to a metabolic disease specialist. -

Anesthetic Considerations and Clinical Manifestations 243
MUCOPOLYSACCHARIDOSES: ANESTHETIC CONSIDERATIONS AND CLINICAL MANIFESTATIONS 243 MUCOPOLYSACCHARIDOSES: ANESTHETIC CONSIDERATIONS AND CLINICAL MANIFESTATIONS * ** *** JERMALE A. SAM , AMIR R. BALUCH , RASHID S. NIAZ , *** **** LINDSEY LONADIER AND ALAN D. KAYE Abstract Mucopolysaccharidosis (MPS) is a group of genetic disorders that presents challenges during anesthetic care and in particular difficulty with airway management. Patients should be managed by experienced anesthesiologists at centers that are familiar with these types of conditions. Rarely encountered disease states have been identified as important topics in the continuing education of clinical anesthesiologists. This review will define MPS, describe the pathophysiology of MPS, describe how patients with this rare lysosomal storage disorders have dysfunction of tissues, cite the incidence of MPS, list the clinical manifestations and specific problems associated with the administration of anesthesia to patients with MPS, present treatment options for patients with MPS, define appropriate preoperative evaluation and perioperative management of these patients, including, to anticipate potential postoperative airway problems. Introduction Mucopolysaccharidoses (MPS) are a group of rare genetic lysosomal storage disorders characterized by the deficiency in or complete lack of necessary lysosomal enzymes required for the stepwise breakdown of glycosaminoglycans (GAGs, also known as mucopolysaccharidoses)1-5. Consequently, fragments of GAGs accumulate intracellularly in the lysosome resulting in cellular enlargement causing disruption/dysfunction of structure and function of tissues. This process leads to numerous clinical abnormalities. Incidence of all types of MPS is reported to be between 1in 10,000 to 1 in 30,000 live births and are transmitted autosomal recessive except for MPS II which is X-linked1,4,5. Pathophysiology Glycosaminoglycans are long-chain complex carbohydrates consisting of repeating sulfated acidic and amino sugar disaccharide units. -

Hurler Syndrome
orphananesthesia Anaesthesia recommendations for patients suffering from Hurler syndrome Disease name: Hurler syndrome ICD 10: E 76.0 Synonyms: Mucopolysaccharidosis (MPS) I-H, Alpha-L-Iduronidase Deficiency; Pfaundler- Hurler Syndrome Hurler Syndrome is a rare lysosomal storage disease belonging to the group of mucopolysaccharidoses Typ I (MPS I) with an autosomal recessive transmission. MPS I is subdivided into three phenotypes of increasing severity: Scheie syndrome being the mildest, Hurler-Scheie syndrome (MPS I-HS) intermediate and Hurler syndrome (MPS I-H) the most severe. The genetic defect in MPS I-H results in a complete deficiency of the enzyme α-L- iduronidase. This leads to progressive accumulation of glycosaminoglycans causing multi- organ dysfunction. Prevalence of MPS I-H is estimated at 0.7-1.6/100 000. Medicine in progress Perhaps new knowledge Every patient is unique Perhaps the diagnostic is wrong Find more information on the disease, its centres of reference and patient organisations on Orphanet: www.orpha.net 1 Disease summary Patients presenting with MPS I-H have progressive cognitive impairment and somatic disorders. The skeletal abnormalities (cervical spine instability, loss of joint range of motion, restricted mobility, growth slowing or arrest in childhood, short stature, typical facial features with a short and stiff neck) seen in these patients are collectively referred to as dysostosis multiplex. Other signs and symptoms include cardiac disease (cardiomyopathies, valvular dysfunction), restrictive lung disease, frequent and recurrent respiratory infections, different types of hernias, communicating hydrocephalus, compression of the spinal cord, corneal clouding, loss of hearing and all types of organomegaly. The airway is obstructed at all levels due to hypertrophy of the soft tissue with narrowed nasal passage, adenotonsillar hypertrophy, macroglossia and thickened laryngeal and pharyngeal structures, subglottic narrowing and tracheobronchomalacia. -

Ocular Features of Treatable Lysosomal Storage Disorders— Fabry Disease, Mucopolysaccharidoses I, II, and VI, and Gaucher Disease
Lanzl.qxp 16/10/08 03:09 Page 44 Anterior Segment Cornea Ocular Features of Treatable Lysosomal Storage Disorders— Fabry Disease, Mucopolysaccharidoses I, II, and VI, and Gaucher Disease a report by Ines M Lanzl, MD, PhD1 and Bart P Leroy, MD, PhD2 1. Department of Ophthalmology, Munich University of Technology; 2. Department of Ophthalmology and Center for Medical Genetics, Ghent University Hospital DOI: 10.17925/USOR.2007.03.00.44 Lysosomal storage disorders (LSDs) are a group of more than 50 are currently treatable with ERT: Fabry disease, mucopolysaccharidosis inheritable disorders.1,2 Although individually rare, they collectively affect (MPS) types I, II, and VI, and Gaucher disease. approximately one in 5,000 live births.3 Defective metabolism of proteins, carbohydrates, or lipids resulting from deficiency of one of the many Fabry Disease lysosomal enzymes leads to pathological accumulation of substances Fabry disease is a life-threatening LSD characterized by a deficient activity of the within the lysosomes. This lysosomal accumulation triggers an insidious lysosomal enzyme α-galactosidase A.9 This deficiency results in a diminished cascade of processes generally leading to progressive tissue damage and ability to catabolize certain glycolipids. Progressive lysosomal accumulation of organ failure. undegraded globotriaosylceramide (GL-3) occurs in many cell types throughout the body, e.g. in vascular endothelial cells, connective tissue, kidneys, heart, Most physicians have difficulty recognizing patients in an early stage of the brain, nerves, and cornea. The initial insult of substrate accumulation leads to disease, as each patient is unique in terms of age of presentation, cluster secondary tissue damage and irreversible organ failure in a cascade of of presenting symptoms, rate of disease progression, and comorbidities. -

Mucopolysaccharidosis I
Mucopolysaccharidosis I Author: Professor Michael Beck1 Creation date: September 2001 Update: September 2003 Scientific editor: Professor Udo Wendel 1 Children`s Hospital, University of Mainz Langenbeckstrasse 1 55101 Mainz. Germany. [email protected] Abstract Keywords Disease names Excluded diseases Diagnostic criteria Differential diagnosis Prevalence Treatment Etiology Diagnostic methods Antenatal diagnosis References Abstract Mucopolysaccharidosis type I (MPS I) is caused by the deficiency of the lysosomal enzyme α-L-iduronidase. In the severe form (Hurler disease) skeletal deformities and a delay in motor and mental development are the leading symptoms. Radiological examination of the skeleton reveals a characteristic pattern that is called dysostosis multiplex. Patients with the adult form (Scheie disease) are of almost normal height and do not show mental retardation. Typical symptoms are stiff joints, corneal opacities, carpal tunnel syndrome and mild skeletal changes. In most cases also the heart is involved in the storage process. Some patients with α-iduronidase deficiency exhibit symptoms that are intermediate between Hurler and Scheie disease. The different phenotypes are caused by allelic mutations of the α-iduronidase (IDUA) gene. Mucopolysaccharidosis I is a rare lysosomal storage disorder, its prevalence being about 1:100 000. Besides palliative treatment bone marrow transplantation can be performed, that, however, is associated with several risks (for example graft-versus-host- reaction). In addition, an enzyme preparation (Laronidase, AldurazymeR) is available that in clinical trials led to improvement of the lung function and joint-mobility, whereby long-time experience is still missing. Keywords mucopolysaccharidosis type I, mucopolysaccharidosis type I-H, mucopolysaccharidosis type I-S, mucopolysaccharidosis type I-HS, Hurler disease, Scheie disease, Hurler/Scheie disease, dysostosis multiplex, Heparan sulphate, Dermatan sulphate, enzyme replacement, gene therapy. -

Ocular Manifestations of Inherited Diseases Maya Eibschitz-Tsimhoni
10 Ocular Manifestations of Inherited Diseases Maya Eibschitz-Tsimhoni ecognizing an ocular abnormality may be the first step in Ridentifying an inherited condition or syndrome. Identifying an inherited condition may corroborate a presumptive diagno- sis, guide subsequent management, provide valuable prognostic information for the patient, and determine if genetic counseling is needed. Syndromes with prominent ocular findings are listed in Table 10-1, along with their alternative names. By no means is this a complete listing. Two-hundred and thirty-five of approxi- mately 1900 syndromes associated with ocular or periocular manifestations (both inherited and noninherited) identified in the medical literature were chosen for this chapter. These syn- dromes were selected on the basis of their frequency, the char- acteristic or unique systemic or ocular findings present, as well as their recognition within the medical literature. The boldfaced terms are discussed further in Table 10-2. Table 10-2 provides a brief overview of the common ocular and systemic findings for these syndromes. The table is organ- ized alphabetically; the boldface name of a syndrome is followed by a common alternative name when appropriate. Next, the Online Mendelian Inheritance in Man (OMIM™) index num- ber is listed. By accessing the OMIM™ website maintained by the National Center for Biotechnology Information at http://www.ncbi.nlm.nih.gov, the reader can supplement the material in the chapter with the latest research available on that syndrome. A MIM number without a prefix means that the mode of inheritance has not been proven. The prefix (*) in front of a MIM number means that the phenotype determined by the gene at a given locus is separate from those represented by other 526 chapter 10: ocular manifestations of inherited diseases 527 asterisked entries and that the mode of inheritance of the phe- notype has been proven. -
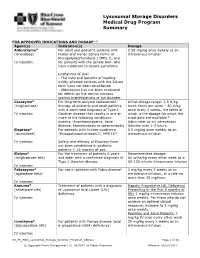
Lysosomal Storage Disorders Medical Drug Program Summary
Lysosomal Storage Disorders Medical Drug Program Summary FDA APPROVED INDICATIONS AND DOSAGE1-11 Agent(s) Indication(s) Dosage Aldurazyme® For adult and pediatric patients with 0.58 mg/kg once weekly as an (laronidase) Hurler and Hurler-Scheie forms of intravenous infusion Mucopolysaccharidosis I (MPS I), and IV injection for patients with the Scheie form who have moderate to severe symptoms Limitations of Use: - The risks and benefits of treating mildly affected patients with the Scheie form have not been established. - Aldurazyme has not been evaluated for effects on the central nervous system manifestations of the disorder. Cerezyme® For long-term enzyme replacement Initial dosage range: 2.5 U/kg (imiglucerase) therapy of pediatric and adult patients three times per week – 60 U/kg with a confirmed diagnosis of Type 1 once every 2 weeks, the latter of IV injection Gaucher disease that results in one or which is the dosage for which the more of the following conditions: most data are available.b anemia, thrombocytopenia, bone Administer as an intravenous disease, hepatomegaly or splenomegaly infusion over 1-2 hours. Elaprase® For patients with Hunter syndrome 0.5 mg/kg once weekly as an (idursulfase) (Mucopolysaccharidosis II, MPS II)a intravenous infusion IV injection Safety and efficacy of Elaprase have not been established in pediatric patients < 16 months of age. Elelyso® For the treatment of patients 4 years Recommended dosage: (taliglucerase alfa) and older with a confirmed diagnosis of 60 units/kg every other week as a Type 1 Gaucher disease 60-120 minute intravenous infusion IV injection Fabrazyme® For use in patients with Fabry diseasec 1 mg/kg every 2 weeks as an (agalsidase beta) intravenous infusion, at a rate no more than 15 mg/hour IV injection Kanuma® Treatment of patients with a diagnosis Rapidly Progressive LAL Deficiency (sebelipase alfa) of Lysosomal Acid Lipase (LAL) Presenting in the first 6 months of deficiency life: 1 mg/kg intravenous infusion IV injection once weekly. -

(Pseudo- Hurler Polydystrophy). a Case Report
OPHTHALMOLOGICAL FINDINGS IN A PATIENT WITH MUCOLIPIDOSIS III (PSEUDO- HURLER POLYDYSTROPHY). A CASE REPORT. POURJAVAN S.*, FRYNS J.P.**, VAN HOVE J.L.K. ***, POORTHUIS B.J.H.M. ****, CASTEELS I.* SUMMARY: stigmatisme. We stellen een patiëntje voor met de bovenbenoemde oftalmologische afwijkingen. Mucolipidosis III (Pseudo- Hurler Polydystrophy) is a rare autosomal recessively inherited Hurler-like dis- KEY-WORDS: ease. The ophthalmological findings in these pa- tients include a triad of mild retinopathy, corneal Pseudo- Hurler polydystrophy, corneal clouding and hyperopic astigmatism. We present a clouding, retinopathy. patient with these ophthalmological characteristics. MOTS-CLÉS: RÉSUMÉ: Pseudo- Hurler polydystrophie, opacités de la La Mucolipidose III (Pseudo- Hurler Polydystrophie) cornée, rétinopathie. est une maladie héréditaire autosomale récessive très rare. Les caractéristiques ophtalmologiques de cet- te maladie consistent en la triade de rétinopathie lé- gère, opacités de la cornée et astigmatisme hypero- pique. Une patiente avec des symptômes ophtalmo- logiques typiques est présentée. SAMENVATTING: Mucolipidosis III ( Pseudo- Hurler Polydystrophy) is een zeldzame autosomaal recessief erfelijke aandoe- ning. De oftalmologische bevindingen bij deze pa- tiënten omvatten een typische triade van milde reti- nopathie, corneale opaciteiten en hypermetroop a- zzzzzz * Department of Ophthalmology, UZ. Leuven, Belgium ** Department of Genetics, UZ.Leuven, Belgium *** Department of Pediatrics, UZ.Leuven, Belgium **** Department of Pediatrics, LUMC, Leiden, The Netherlands received: 08.07.02 accepted: 20.08.02 Bull. Soc. belge Ophtalmol., 286, 19-24, 2002. 19 INTRODUCTION CASE REPORT Mucolipidosis II (I-cell disease, Leroy disease) A thirteen-year-old girl with the clinical char- and III (ML-III or pseudo-Hurler polydystrophy) acteristics of ML-III presented for ophthalmo- are respectively the severe and mild forms of logical evaluation as a part of a multidisci- an autosomal recessive genetic disorder char- plinary investigation. -

Mucopolysaccharidosis, Type I (MPSI)
Mucopolysaccharidosis type I Also known as: • MPS I • Hurler-Scheie syndrome • Hurler syndrome • IDUA deficiency • MPS I H • MPS I H-S • MPS I S • Scheie syndrome Definition: The IDUA gene provides instructions to produce the alpha-L-iduronidase (IDUA) enzyme that breaks down large sugar molecules in the body, which are called glycosaminoglycans. Mutations in the IDUA gene reduce or eliminate the function of the IDUA enzyme, which leads to a dangerous buildup of glycosaminoglycans within cells, specifically the lysosomes. The buildup of glycosaminoglycans increases the size of the lysosomes, which can lead to enlarged tissues/organs and cause a variety of symptoms. This buildup may also interfere with the function of some proteins inside the lysosomes, and disrupt the movement of molecules within the cell. There are two main types of MPS I, which are referred to as severe MPS I and attenuated MPS I. Diagnosis: Sequencing of the IDUA gene will be performed as part of the screening algorithm. Diagnostic confirmation, under the direction of a specialist, may include measurement of glycosaminoglycans in urine and IDUA enzyme activity in blood. How is it inherited: MPS I is inherited in an autosomal recessive pattern. Normally, a person has two functional copies of the IDUA gene. In people with MPS I, both copies of the gene have a mutation. Each parent of a newborn with MPS I typically has one functional gene and one mutated gene, and is considered a carrier. When both parents are carriers, the chance of a newborn inheriting two mutated genes is 25%. Newborn Screening: • Incidence: The estimated incidence of severe MPS I in the general population is 1 in 100,000, while the incidence of attenuated (less severe) MPS is 1 in 500,000.