Basic Elasticity and Viscoelasticity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

10-1 CHAPTER 10 DEFORMATION 10.1 Stress-Strain Diagrams And
EN380 Naval Materials Science and Engineering Course Notes, U.S. Naval Academy CHAPTER 10 DEFORMATION 10.1 Stress-Strain Diagrams and Material Behavior 10.2 Material Characteristics 10.3 Elastic-Plastic Response of Metals 10.4 True stress and strain measures 10.5 Yielding of a Ductile Metal under a General Stress State - Mises Yield Condition. 10.6 Maximum shear stress condition 10.7 Creep Consider the bar in figure 1 subjected to a simple tension loading F. Figure 1: Bar in Tension Engineering Stress () is the quotient of load (F) and area (A). The units of stress are normally pounds per square inch (psi). = F A where: is the stress (psi) F is the force that is loading the object (lb) A is the cross sectional area of the object (in2) When stress is applied to a material, the material will deform. Elongation is defined as the difference between loaded and unloaded length ∆푙 = L - Lo where: ∆푙 is the elongation (ft) L is the loaded length of the cable (ft) Lo is the unloaded (original) length of the cable (ft) 10-1 EN380 Naval Materials Science and Engineering Course Notes, U.S. Naval Academy Strain is the concept used to compare the elongation of a material to its original, undeformed length. Strain () is the quotient of elongation (e) and original length (L0). Engineering Strain has no units but is often given the units of in/in or ft/ft. ∆푙 휀 = 퐿 where: is the strain in the cable (ft/ft) ∆푙 is the elongation (ft) Lo is the unloaded (original) length of the cable (ft) Example Find the strain in a 75 foot cable experiencing an elongation of one inch. -

Mechanics of Materials Plasticity
Provided for non-commercial research and educational use. Not for reproduction, distribution or commercial use. This article was originally published in the Reference Module in Materials Science and Materials Engineering, published by Elsevier, and the attached copy is provided by Elsevier for the author’s benefit and for the benefit of the author’s institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who you know, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier’s permissions site at: http://www.elsevier.com/locate/permissionusematerial Lubarda V.A., Mechanics of Materials: Plasticity. In: Saleem Hashmi (editor-in-chief), Reference Module in Materials Science and Materials Engineering. Oxford: Elsevier; 2016. pp. 1-24. ISBN: 978-0-12-803581-8 Copyright © 2016 Elsevier Inc. unless otherwise stated. All rights reserved. Author's personal copy Mechanics of Materials: Plasticity$ VA Lubarda, University of California, San Diego, CA, USA r 2016 Elsevier Inc. All rights reserved. 1 Yield Surface 1 1.1 Yield Surface in Strain Space 2 1.2 Yield Surface in Stress Space 2 2 Plasticity Postulates, Normality and Convexity -

Identification of Rock Mass Properties in Elasto-Plasticity D
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Identification of rock mass properties in elasto-plasticity D. Deng, D. Nguyen Minh To cite this version: D. Deng, D. Nguyen Minh. Identification of rock mass properties in elasto-plasticity. Computers and Geotechnics, Elsevier, 2003, 30, pp.27-40. 10.1016/S0266-352X(02)00033-2. hal-00111371 HAL Id: hal-00111371 https://hal.archives-ouvertes.fr/hal-00111371 Submitted on 26 Sep 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Identification of rock mass properties in elasto-plasticity Desheng Deng*, Duc Nguyen-Minh Laboratoire de Me´canique des Solides, E´cole Polytechnique, 91128 Palaiseau, France Abstract A simple and effective back analysis method has been proposed on the basis of a new cri-terion of identification, the minimization of error on the virtual work principle. This method works for both linear elastic and nonlinear elasto-plastic problems. The elasto-plastic rock mass properties for different criteria of plasticity can be well identified based on field measurements. -

Lecture 13: Earth Materials
Earth Materials Lecture 13 Earth Materials GNH7/GG09/GEOL4002 EARTHQUAKE SEISMOLOGY AND EARTHQUAKE HAZARD Hooke’s law of elasticity Force Extension = E × Area Length Hooke’s law σn = E εn where E is material constant, the Young’s Modulus Units are force/area – N/m2 or Pa Robert Hooke (1635-1703) was a virtuoso scientist contributing to geology, σ = C ε palaeontology, biology as well as mechanics ij ijkl kl ß Constitutive equations These are relationships between forces and deformation in a continuum, which define the material behaviour. GNH7/GG09/GEOL4002 EARTHQUAKE SEISMOLOGY AND EARTHQUAKE HAZARD Shear modulus and bulk modulus Young’s or stiffness modulus: σ n = Eε n Shear or rigidity modulus: σ S = Gε S = µε s Bulk modulus (1/compressibility): Mt Shasta andesite − P = Kεv Can write the bulk modulus in terms of the Lamé parameters λ, µ: K = λ + 2µ/3 and write Hooke’s law as: σ = (λ +2µ) ε GNH7/GG09/GEOL4002 EARTHQUAKE SEISMOLOGY AND EARTHQUAKE HAZARD Young’s Modulus or stiffness modulus Young’s Modulus or stiffness modulus: σ n = Eε n Interatomic force Interatomic distance GNH7/GG09/GEOL4002 EARTHQUAKE SEISMOLOGY AND EARTHQUAKE HAZARD Shear Modulus or rigidity modulus Shear modulus or stiffness modulus: σ s = Gε s Interatomic force Interatomic distance GNH7/GG09/GEOL4002 EARTHQUAKE SEISMOLOGY AND EARTHQUAKE HAZARD Hooke’s Law σij and εkl are second-rank tensors so Cijkl is a fourth-rank tensor. For a general, anisotropic material there are 21 independent elastic moduli. In the isotropic case this tensor reduces to just two independent elastic constants, λ and µ. -

Crystal Plasticity Model with Back Stress Evolution. Wei Huang Louisiana State University and Agricultural & Mechanical College
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1996 Crystal Plasticity Model With Back Stress Evolution. Wei Huang Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Huang, Wei, "Crystal Plasticity Model With Back Stress Evolution." (1996). LSU Historical Dissertations and Theses. 6154. https://digitalcommons.lsu.edu/gradschool_disstheses/6154 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type o f computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Cohesive Crack with Rate-Dependent Opening and Viscoelasticity: I
International Journal of Fracture 86: 247±265, 1997. c 1997 Kluwer Academic Publishers. Printed in the Netherlands. Cohesive crack with rate-dependent opening and viscoelasticity: I. mathematical model and scaling ZDENEKÏ P. BAZANTÏ and YUAN-NENG LI Department of Civil Engineering, Northwestern University, Evanston, Illinois 60208, USA Received 14 October 1996; accepted in revised form 18 August 1997 Abstract. The time dependence of fracture has two sources: (1) the viscoelasticity of material behavior in the bulk of the structure, and (2) the rate process of the breakage of bonds in the fracture process zone which causes the softening law for the crack opening to be rate-dependent. The objective of this study is to clarify the differences between these two in¯uences and their role in the size effect on the nominal strength of stucture. Previously developed theories of time-dependent cohesive crack growth in a viscoelastic material with or without aging are extended to a general compliance formulation of the cohesive crack model applicable to structures such as concrete structures, in which the fracture process zone (cohesive zone) is large, i.e., cannot be neglected in comparison to the structure dimensions. To deal with a large process zone interacting with the structure boundaries, a boundary integral formulation of the cohesive crack model in terms of the compliance functions for loads applied anywhere on the crack surfaces is introduced. Since an unopened cohesive crack (crack of zero width) transmits stresses and is equivalent to no crack at all, it is assumed that at the outset there exists such a crack, extending along the entire future crack path (which must be known). -

An Investigation Into the Effects of Viscoelasticity on Cavitation Bubble Dynamics with Applications to Biomedicine
An Investigation into the Effects of Viscoelasticity on Cavitation Bubble Dynamics with Applications to Biomedicine Michael Jason Walters School of Mathematics Cardiff University A thesis submitted for the degree of Doctor of Philosophy 9th April 2015 Summary In this thesis, the dynamics of microbubbles in viscoelastic fluids are investigated nu- merically. By neglecting the bulk viscosity of the fluid, the viscoelastic effects can be introduced through a boundary condition at the bubble surface thus alleviating the need to calculate stresses within the fluid. Assuming the surrounding fluid is incompressible and irrotational, the Rayleigh-Plesset equation is solved to give the motion of a spherically symmetric bubble. For a freely oscillating spherical bubble, the fluid viscosity is shown to dampen oscillations for both a linear Jeffreys and an Oldroyd-B fluid. This model is also modified to consider a spherical encapsulated microbubble (EMB). The fluid rheology affects an EMB in a similar manner to a cavitation bubble, albeit on a smaller scale. To model a cavity near a rigid wall, a new, non-singular formulation of the boundary element method is presented. The non-singular formulation is shown to be significantly more stable than the standard formulation. It is found that the fluid rheology often inhibits the formation of a liquid jet but that the dynamics are governed by a compe- tition between viscous, elastic and inertial forces as well as surface tension. Interesting behaviour such as cusping is observed in some cases. The non-singular boundary element method is also extended to model the bubble tran- sitioning to a toroidal form. -
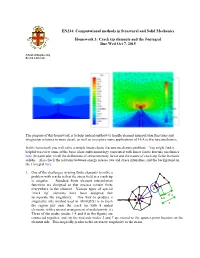
Crack Tip Elements and the J Integral
EN234: Computational methods in Structural and Solid Mechanics Homework 3: Crack tip elements and the J-integral Due Wed Oct 7, 2015 School of Engineering Brown University The purpose of this homework is to help understand how to handle element interpolation functions and integration schemes in more detail, as well as to explore some applications of FEA to fracture mechanics. In this homework you will solve a simple linear elastic fracture mechanics problem. You might find it helpful to review some of the basic ideas and terminology associated with linear elastic fracture mechanics here (in particular, recall the definitions of stress intensity factor and the nature of crack-tip fields in elastic solids). Also check the relations between energy release rate and stress intensities, and the background on the J integral here. 1. One of the challenges in using finite elements to solve a problem with cracks is that the stress field at a crack tip is singular. Standard finite element interpolation functions are designed so that stresses remain finite a everywhere in the element. Various types of special b c ‘crack tip’ elements have been designed that 3L/4 incorporate the singularity. One way to produce a L/4 singularity (the method used in ABAQUS) is to mesh L the region just near the crack tip with 8 noded elements, with a special arrangement of nodal points: (i) Three of the nodes (nodes 1,4 and 8 in the figure) are connected together, and (ii) the mid-side nodes 2 and 7 are moved to the quarter-point location on the element side. -

Analysis of Viscoelastic Flexible Pavements
Analysis of Viscoelastic Flexible Pavements KARL S. PISTER and CARL L. MONISMITH, Associate Professor and Assistant Professor of Civil Engineering, University of California, Berkeley To develop a better understandii^ of flexible pavement behavior it is believed that components of the pavement structure should be considered to be viscoelastic rather than elastic materials. In this paper, a step in this direction is taken by considering the asphalt concrete surface course as a viscoelastic plate on an elastic foundation. Assumptions underlying existing stress and de• formation analyses of pavements are examined. Rep• resentations of the flexible pavement structure, im• pressed wheel loads, and mechanical properties of as• phalt concrete and base materials are discussed. Re• cent results pointing toward the viscoelastic behavior of asphaltic mixtures are presented, including effects of strain rate and temperature. IJsii^ a simplified viscoelastic model for the asphalt concrete surface course, solutions for typical loading problems are given. • THE THEORY of viscoelasticity is concerned with the behavior of materials which exhibit time-dependent stress-strain characteristics. The principles of viscoelasticity have been successfully used to explain the mechanical behavior of high polymers and much basic work U) lias been developed in this area. In recent years the theory of viscoelasticity has been employed to explain the mechanical behavior of asphalts (2, 3, 4) and to a very limited degree the behavior of asphaltic mixtures (5, 6, 7) and soils (8). Because these materials have time-dependent stress-strain characteristics and because they comprise the flexible pavement section, it seems reasonable to analyze the flexible pavement structure using viscoelastic principles. -

Engineering Viscoelasticity
ENGINEERING VISCOELASTICITY David Roylance Department of Materials Science and Engineering Massachusetts Institute of Technology Cambridge, MA 02139 October 24, 2001 1 Introduction This document is intended to outline an important aspect of the mechanical response of polymers and polymer-matrix composites: the field of linear viscoelasticity. The topics included here are aimed at providing an instructional introduction to this large and elegant subject, and should not be taken as a thorough or comprehensive treatment. The references appearing either as footnotes to the text or listed separately at the end of the notes should be consulted for more thorough coverage. Viscoelastic response is often used as a probe in polymer science, since it is sensitive to the material’s chemistry and microstructure. The concepts and techniques presented here are important for this purpose, but the principal objective of this document is to demonstrate how linear viscoelasticity can be incorporated into the general theory of mechanics of materials, so that structures containing viscoelastic components can be designed and analyzed. While not all polymers are viscoelastic to any important practical extent, and even fewer are linearly viscoelastic1, this theory provides a usable engineering approximation for many applications in polymer and composites engineering. Even in instances requiring more elaborate treatments, the linear viscoelastic theory is a useful starting point. 2 Molecular Mechanisms When subjected to an applied stress, polymers may deform by either or both of two fundamen- tally different atomistic mechanisms. The lengths and angles of the chemical bonds connecting the atoms may distort, moving the atoms to new positions of greater internal energy. -

Elastic Properties of Fe−C and Fe−N Martensites
Elastic properties of Fe−C and Fe−N martensites SOUISSI Maaouia a, b and NUMAKURA Hiroshi a, b * a Department of Materials Science, Osaka Prefecture University, Naka-ku, Sakai 599-8531, Japan b JST-CREST, Gobancho 7, Chiyoda-ku, Tokyo 102-0076, Japan * Corresponding author. E-mail: [email protected] Postal address: Department of Materials Science, Graduate School of Engineering, Osaka Prefecture University, Gakuen-cho 1-1, Naka-ku, Sakai 599-8531, Japan Telephone: +81 72 254 9310 Fax: +81 72 254 9912 1 / 40 SYNOPSIS Single-crystal elastic constants of bcc iron and bct Fe–C and Fe–N alloys (martensites) have been evaluated by ab initio calculations based on the density-functional theory. The energy of a strained crystal has been computed using the supercell method at several values of the strain intensity, and the stiffness coefficient has been determined from the slope of the energy versus square-of-strain relation. Some of the third-order elastic constants have also been evaluated. The absolute magnitudes of the calculated values for bcc iron are in fair agreement with experiment, including the third-order constants, although the computed elastic anisotropy is much weaker than measured. The tetragonally distorted dilute Fe–C and Fe–N alloys exhibit lower stiffness than bcc iron, particularly in the tensor component C33, while the elastic anisotropy is virtually the same. Average values of elastic moduli for polycrystalline aggregates are also computed. Young’s modulus and the rigidity modulus, as well as the bulk modulus, are decreased by about 10 % by the addition of C or N to 3.7 atomic per cent, which agrees with the experimental data for Fe–C martensite. -

Viscoelasticity of Filled Elastomers: Determination of Surface-Immobilized Components and Their Role in the Reinforcement of SBR-Silica Nanocomposites
Viscoelasticity of Filled Elastomers: Determination of Surface-Immobilized Components and their Role in the Reinforcement of SBR-Silica Nanocomposites Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) genehmigt durch die Naturwissenschaftliche Fakultät II Institut für Physik der Martin-Luther-Universität Halle-Wittenberg vorgelegt von M. Sc. Anas Mujtaba geboren am 03.04.1980 in Lahore, Pakistan Halle (Saale), January 15th, 2014 Gutachter: 1. Prof. Dr. Thomas Thurn-Albrecht 2. Prof. Dr. Manfred Klüppel 3. Prof. Dr. Rene Androsch Öffentliche Verteidigung: July 3rd, 2014 In loving memory of my beloved Sister Rabbia “She will be in my Heart ξ by my Side for the Rest of my Life” Contents 1 Introduction 1 2 Theoretical Background 5 2.1 Elastomers . .5 2.1.1 Fundamental Theories on Rubber Elasticity . .7 2.2 Fillers . 10 2.2.1 Carbon Black . 12 2.2.2 Silica . 12 2.3 Filled Rubber Reinforcement . 15 2.3.1 Occluded Rubber . 16 2.3.2 Payne Effect . 17 2.3.3 The Kraus Model for the Strain-Softening Effect . 18 2.3.4 Filler Network Reinforcement . 20 3 Experimental Methods 29 3.1 Dynamic Mechanical Analysis . 29 3.1.1 Temperature-dependent Measurement (Temperature Sweeps) . 31 3.1.2 Time-Temperature Superposition (Master Curves) . 31 3.1.3 Strain-dependent Measurement (Payne Effect) . 33 3.2 Low-field NMR . 34 3.2.1 Theoretical Concept . 34 3.2.2 Experimental Details . 39 4 Optimizing the Tire Tread 43 4.1 Relation Between Friction and the Mechanical Properties of Tire Rubbers 46 4.2 Usage of tan δ As Loss Parameter .