The Genetics of Monarch Butterfly Migration and Warning Colouration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Letters from a Fish to His Admiral (Pdf)
LETTERS FROM A FISH TO HIS ADMIRAL BEING BRITANNIA NEWSLETTERS 1-20 17th November 1953 – 9th April 1954 from Acting Captain J S Dalglish, Royal Navy to Rear Admiral Connoly Abel Smith, Royal Navy (temporarily absent with Her Majesty Queen Elizabeth II on board the S.S. GOTHIC for the Commonwealth tour of 1953/4) COMMISSIONING BRITANNIA The following notes and letters were written by Acting Captain J S Dalglish, the Officer in charge of Commissioning BRITANNIA, and getting her ready for Royal service, between November 1953 and April 1954. They were written to Rear Admiral Conolly Abel Smith who was on SS Gothic, in temporary use as a Royal Yacht, for The Queen’s Commonwealth Tour 53/54. Upon his return to the UK, Admiral Abel Smith would then become in charge of BRITANNIA, as the first Flag Officer Royal Yachts (FORY). Throughout his career Captain Dalglish was known as FISH. We have a copy of his autobiography – Life Story of a Fish. Prologue SOME RANDOM THOUGHTS ON “BRITANNIA” AT THE BEGINNING. Selection Officers were nominated by Flag Officer Royal Yachts after personal interviews, after which approval was formally given from the Palace. Royal Yachtsmen (who were ranked as ‘Riggers’ in V&A, but with us they became just ‘Yachtsmen’), were all volunteers in response to an AFO in 1953. About 1,000 names came up and we began, of course, with examination of Service Certificates from which any who were not VG Superior were eliminated (although I seem to remember some of our Stokers were VG Satisfactory because of numbers). -

La Reserva Natural Del Puerto Mar Del Plata, Oasis Urbano De Vida Silvestre
RESERVA NATURAL DEL PUERTO MAR DEL PLATA, UN OASIS URBANO DE VIDA SILVESTRE Universidad FASTA Gran Canciller Dr. Fr. Aníbal E. Fosbery Rector Dr. Juan Carlos Mena Decano Facultad de Ingeniería Ing. Roberto Giordano Lerena Secretaria de Investigación y Postgrado Lic. Mónica Pascual Coordinador de Ingeniería Ambiental Ing. Simón Caparrós RESERVA NATURAL DEL PUERTO MAR DEL PLATA, UN OASIS URBANO DE VIDA SILVESTRE Silvia Graciela De Marco Laura Estela Vega Patricio Juan Bellagamba Universidad FASTA Mar del Plata, junio de 2011 2010 -2016 Bicentenario de la Patria Reserva Natural del Puerto Mar del Plata, un oasis urbano de vida silvestre / coordinado por Silvia Graciela De Marco ; Laura Estela Vega ; Patricio Juan Bellagamba. - 1a ed. - Mar del Plata : Universidad FASTA, 2011. Ebook . : il. eISBN 978-987-1312-37-5 1. Reservas Naturales. I. De Marco, Silvia Graciela, coord. II. Vega, Laura Estela, coord. III. Bellagamba, Patricio Juan, coord. CDD 333.951 6 Fecha de catalogación: 02/06/2010 Responsable de Edición Lic. José Miguel Ravasi © Universidad FASTA Ediciones Facultad de Ingeniería. Universidad FASTA. Gascón 3145 – B7600FNK Mar del Plata, Argentina +54 223 4990400 [email protected]; [email protected] edición digital junio de 2011 Queda hecho el depósito que establece la Ley 11.723 Impreso en Argentina - Printed in Argentina Queda prohibida, salvo excepción prevista en la ley, cualquier forma de reproducción, distribución, comunicación pública y transformación de esta obra sin contar con la autorización de los titulares -

An Updated List of the Butterflies of Chile (Lepidoptera
9 Boletín del Museo Nacional de Historia Natural, Chile, 63: 9-31 (2014) AN UPDATED LIST OF THE BUTTERFLIES OF CHILE (LEPIDOPTERA, PAPILIONOIDEA AND HESPERIOIDEA) INCLUDING DISTRIBUTION, FLIGHT PERIOD AND CONSERVATION STATUS PART I, COMPRISING THE FAMILIES: PAPILIONIDAE, PIERIDAE, NYM- PHALIDAE (IN PART) AND HESPERIIDAE DESCRIBING A NEW SPECIES OF HYPSOCHILA (PIERIDAE) AND A NEW SUBSPECIES OF YRAMEA MODESTA (NYMPHALIDAE) Dubi Benyamini1, Alfredo Ugarte2, Arthur M. Shapiro3, Olaf Hermann Hendrik Mielke4, Tomasz Pyrcz 5 and Zsolt Bálint6 1 4D MicroRobotics, Israel [email protected]; 2 P. O. Box 2974, Santiago, Chile augartepena@ gmail.com; 3 Center for Population Biology, University of California, Davis, CA 95616, U.S.A. amsha- [email protected]; 4 University of Parana, Brazil [email protected]; 5 Zoological Museum, Jagiellonian University, Krakow, Poland [email protected]; 6 Hungarian National History Museum, Budapest, Hungary. [email protected] ABSTRACT During more than half a century, Luis Peña and later his collaborator Alfredo Ugarte, gathered all known butterfl y data and suspected Chilean specimens to publish their seminal book on the butterfl ies of Chile (Peña and Ugarte 1997). Their work summarized the accumulated knowledge up to the end of the 20th century. Since then much additional work has been done by the authors, resulting in the descriptions of numerous new species as well as establishing new species records for Chile, especially in the families Lycaenidae and Nymphalidae (Satyrinae). The list of these two families is still not com- plete, as several new species will be published soon and will appear in part II of this paper. The present work involving four families updates the Chilean list by: 1) describing one new species of Pieridae, 2) describing one new subspecies of Nymphalidae (Heliconiinae), 3) adding in total 10 species and two subspecies to the Chilean list. -

Migration and Host Plant Use of the Southern Monarch, Danaus Erippus
Migration and host plant use of the southern monarch, Danaus erippus Stephen Malcolm and Benjamin Slager Department of Biological Sciences, Western Michigan University Slide 1 Field research: 1. Bolivia: (a) Tropical lowlands near Santa Cruz and Buena Vista east to Santiago de Chiquitos. (b) Andean highlands from Sucre south to Potosí and Tarija 2. Argentina: (a) Eastern cordillera of the Andes from Tucumán north to Salta and Jujuy. (b) Eastern Andean valleys along the edge of Yungas ecosystem Slide 2 Data from museum collections: 1) Dr G. Lamas, Universidad Mayor de San Marcos, Lima 2) BMNH, London 3) McGuire Center for Lepidoptera and Biodiversity, Gainesville, FL. Danaus plexippus nigrippus and Danaus erippus Slide 3 Distribution of Danaus erippus from museum records Slide 4 Published information • Sequence of 6 papers from K.J. Hayward in Argentina: – Records include spring, summer and autumn seasons – D. erippus only appears in the records in April and May (autumn). – Approx. 30 days of observations: • D. erippus was observed moving south in all of them except for two observations on 13 April 1928 and 30 April 1970 when seen flying north. – Hayward (1970) says northward flight was unusual & D. erippus is always seen flying south in the autumn parallel to the Andes: • Hayward, K.J. 1928. The Entomologist 61: 210-212. • Hayward, K.J. 1962. The Entomologist 95: 8-12. • Hayward, K.J. 1963. The Entomologist 96: 258-264. • Hayward, K.J. 1964. The Entomologist 97: 272-273. • Hayward, K.J. 1967. The Entomologist 100: 29-34. • Hayward, K.J. 1972. The Entomologist 105: 206-208. -

“Subspecies” of New World Monarch Butterflies
CONSERVATION BIOLOGY AND BIODIVERSITY Geographical Populations and “Subspecies” of New World Monarch Butterflies (Nymphalidae) Share a Recent Origin and Are Not Phylogenetically Distinct 1, 2 1, 3 ANDREW V. Z. BROWER AND MIRANDA M. JEANSONNE Ann. Entomol. Soc. Am. 97(3): 519Ð523 (2004) ABSTRACT To test prior results with a more sensitive technique and larger sample sizes, we assessed genetic diversity among far-ßung monarch butterßy, Danaus plexippus (L.) (Lepidoptera: Nympha- lidae), populations from North and South America by using mitochondrial DNA sequences. Our new data corroborate the previously documented close genetic similarity among individuals and reveal no phylogenetic structure among populations throughout the speciesÕ New World range in North and South America. Despite this intraspeciÞc homogeneity, the monarch is clearly distinct from its sister taxon Danaus erippus (Cramer). The evidence suggests that the monarch has colonized its current distribution in relatively recent evolutionary time. Implications for conservation and regulatory policy over interregional transfer are discussed. KEY WORDS mtDNA, Danaus plexippus, Nymphalidae, biogeography, genetic diversity THE MONARCH BUTTERFLY, Danaus plexippus (L.), is per- temperate North America during the summer haps the worldÕs most charismatic butterßy species, (Brower 1977). famed for its milkweed-derived chemical defense, its As noted, migratory monarch populations overwin- involvement in mimicry, and most notably its spec- ter in two separate regions of North America, and tacular annual migratory cycle and overwintering ag- there has been considerable discussion in the litera- gregations of the North American populations on the ture as to whether there has been mixing of popula- California coast and in montane central Mexico. The tions east and west of the Rocky Mountains (reviewed distribution of the monarch ranges from southern in Brower 1995). -

The Specialization Continuum Decision-Making in Butterflies With
Behavioural Processes 165 (2019) 14–22 Contents lists available at ScienceDirect Behavioural Processes journal homepage: www.elsevier.com/locate/behavproc The specialization continuum: Decision-making in butterflies with different diet requirements T ⁎ Bruna de Cássia Menezes Ramosa, , José Roberto Trigob, Daniela Rodriguesa a Laboratório de Interações Inseto-Planta and Programa de Pós-Graduação em Ecologia, Instituto de Biologia, Universidade Federal do Rio de Janeiro, PO Box 68020. 21941-902. Cidade Universitária, Ilha do Fundão, Rio de Janeiro, RJ, Brazil b Departamento de Biologia Animal, Instituto de Biologia, Universidade Estadual de Campinas, PO Box 6109, 13083-970, Campinas, SP, Brazil ARTICLE INFO ABSTRACT Keywords: Differences in diet requirements may be reflected in how floral visitors make decisions when probing nectar Agraulis sources that differ in chemical composition. We examined decision-making in butterflies that form a speciali- Danaus zation continuum involving pyrrolizidine alkaloids (PAs) when interacting with PA and non-PA plants: Agraulis Cognition vanillae (non-specialist), Danaus erippus (low demanding PA-specialist) and D. gilippus (high demanding PA- Pyrrolizidine alkaloids specialist). In addition, we assessed whether experience affected decision-making. Butterflies were tested on Toxic nectar either Tridax procumbens (absence of PAs in nectar) or Ageratum conyzoides flowers (presence of PAs in nectar). Agraulis vanillae showed more acceptance for T. procumbens and more rejection for A. conyzoides;nodifferences were recorded for both Danaus species. Agraulis vanillae fed less on A. conyzoides than both Danaus species, which did not differ in this regard. In all butterfly species, experience on PA flowers did not affect feeding time. In the field, butterflies rarely visited PA flowers, regardless of the specialization degree. -

Petition to Protect the Monarch Butterfly (Danaus Plexippus Plexippus) Under the Endangered Species Act
BEFORE THE SECRETARY OF THE INTERIOR © Jeffrey E. Belth PETITION TO PROTECT THE MONARCH BUTTERFLY (DANAUS PLEXIPPUS PLEXIPPUS) UNDER THE ENDANGERED SPECIES ACT Notice of Petition Sally Jewell, Secretary U.S. Department of the Interior 1849 C Street NW Washington, D.C. 20240 [email protected] Dan Ashe, Director U.S. Fish and Wildlife Service 1849 C Street NW Washington, D.C. 20240 [email protected] Douglas Krofta, Chief Branch of Listing, Endangered Species Program U.S. Fish and Wildlife Service 4401 North Fairfax Drive, Room 420 Arlington, VA 22203 [email protected] Monarch ESA Petition 2 PETITIONERS The Center for Biological Diversity (“Center”) is a nonprofit, public interest environmental organization dedicated to the protection of imperiled species and the habitat and climate they need to survive through science, policy, law, and creative media. The Center is supported by more than 775,000 members and activists throughout the country. The Center works to secure a future for all species, great or small, hovering on the brink of extinction. The Center for Food Safety (“CFS”) is a nonprofit public interest organization established in 1997 whose mission centers on protecting public health and the environment by curbing the adverse impacts of industrial agriculture and food production systems on public health, the environment, and animal welfare, and by instead promoting sustainable forms of agriculture. As particularly relevant here, CFS is the leading nonprofit working on the adverse impacts of genetically engineered crops and neonicotinoid pesticides. CFS and its over half-a-million members are concerned about the impacts of industrial agriculture on biodiversity generally, and on monarch butterflies specifically. -

Telling Pacific Lives
TELLING PACIFIC LIVES PRISMS OF PROCESS TELLING PACIFIC LIVES PRISMS OF PROCESS Brij V. Lal & Vicki Luker Editors Published by ANU E Press The Australian National University Canberra ACT 0200, Australia Email: [email protected] This title is also available online at: http://epress.anu.edu.au/tpl_citation.html National Library of Australia Cataloguing-in-Publication entry Title: Telling Pacific lives : prisms of process / editors, Vicki Luker ; Brij V. Lal. ISBN: 9781921313813 (pbk.) 9781921313820 (pdf) Notes: Includes index. Subjects: Islands of the Pacific--Biography. Islands of the Pacific--Anecdotes. Islands of the Pacific--Civilization. Islands of the Pacific--Social life and customs. Other Authors/Contributors: Luker, Vicki. Lal, Brij. Dewey Number: 990.0099 All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying or otherwise, without the prior permission of the publisher. Cover design by Teresa Prowse Cover image: Choris, Louis, 1795-1828. Iles Radak [picture] [Paris : s.n., [1827] 1 print : lithograph, hand col.; 20.5 x 26 cm. nla.pic-an10412525 National Library of Australia Printed by University Printing Services, ANU This edition © 2008 ANU E Press Table of Contents Preface vii 1. Telling Pacic Lives: From Archetype to Icon, Niel Gunson 1 2. The Kila Wari Stories: Framing a Life and Preserving a Cosmology, Deborah Van Heekeren 15 3. From ‘My Story’ to ‘The Story of Myself’—Colonial Transformations of Personal Narratives among the Motu-Koita of Papua New Guinea, Michael Goddard 35 4. Mobility, Modernisation and Agency: The Life Story of John Kikang from Papua New Guinea, Wolfgang Kempf 51 5. -

A Classification of Danaus Butterflies (Lepidoptera: Nymphalidae)
Blackwell Science, LtdOxford, UKZOJZoological Journal of the Linnean Society0024-4082The Lin- nean Society of London, 2005? 2005 1442 191212 Original Article CLASSIFICATION OF DANAUSD. A. S. SMITH ET AL. Zoological Journal of the Linnean Society, 2005, 144, 191–212. With 3 figures A classification of Danaus butterflies (Lepidoptera: Nymphalidae) based upon data from morphology and DNA DAVID A. S. SMITH FLS1*, GUGS LUSHAI FLS2 and JOHN A. ALLEN FLS2 1Natural History Museum, Eton College, Windsor SL4 6EW, UK 2Ecology & Biodiversity Division, School of Biological Sciences, University of Southampton SO16 7PX, UK Received September 2003; accepted for publication March 2005 Classification of the cosmopolitan butterfly genus Danaus (Nymphalidae: Danainae) is revised at subgeneric, specific and subspecific levels, combining for the first time mitochondrial and nuclear DNA sequence information with mor- phological data. Tree topologies based on the nuclear genome (allozymes, pheromone components, the morphology of all life history stages and nuclear DNA sequences), on the one hand, and mitochondrial DNA, on the other, are incon- gruent and challenge the current taxonomy of the genus. Although earlier classifications, based on adult morphology alone, are, in general, well supported by an analysis of total evidence, the mitochondrial phylogeny shows that the species D. chrysippus and its subgenus Anosia are deeply paraphyletic. Subspecies dorippus of D. chrysippus is the basal clade of the genus and is reinstated as the species D. dorippus. The former species D. plexaure is demoted to a subspecies of D. eresimus. The specific status of D. erippus, as distinct from D. plexippus, is tentatively supported. On the strength of the new data, division of the monophyletic genus Danaus s.l. -

Sequential Partial Migration Across Monarch Generations in Michigan
Anim. Migr. 2018; 5: 104-114 Research Article Open Access Natalia Ruiz Vargas, Logan Rowe, Joel Stevens, Joshua E. Armagost, Andrew C. Johnson, Stephen B. Malcolm* Sequential Partial Migration Across Monarch Generations in Michigan https://doi.org/10.1515/ami-2018-0007 Keywords: monarch butterfly, partial migration, voltinism, Received June 13, 2018; accepted October 30, 2018 milkweed hostplant, wing wear, wing load, fat content, cardenolide sequestration Abstract: Running title: Monarch alternative migration Abstract: We collected 434 adult monarchs and surveyed milkweeds for immature monarchs in southwest Michigan, 1 Introduction USA in order to test the hypothesis that monarchs are temporally variable, sequential partial migrants rather Populations of many mobile vertebrate species such as than partial migrants that may be spatially separated. birds, mammals and fish with multi-year lifespans show Adult size, wing wear, female egg counts, fat content mixtures of migratory and non-migratory, or resident life and sequestered chemical defenses were measured in histories characteristic of partial migration [1-3]. Chapman monarchs across an entire season from spring migrant et al., [2] and Shaw and Levin [3] describe three forms arrival, through breeding, until autumn migrant departure. of seasonal partial migrants in which (1) migrants and We predicted that a population characterized by starting residents breed together, but overwinter apart, (2) migrants from all migrants and no residents, through breeding and residents breed apart and overwinter together, or (3) residents, to all migrants and no residents should show migrants breed together, but not every year, generating life history measures consistent with changes in these a mix of migrants and non-migrants. -

Stanton Marianaalves M.Pdf
ii iii Agradecimentos Gostaria de agradecer a todos que me ajudaram e incentivaram ao longo desse trabalho: Em primeiro lugar, aos meus pais, Virgilia e Michael, por me apoiarem em todos os momentos, pelos conselhos e consolos. Aos meus irmãos, Leonardo e Anthony, que mesmo de longe torceram sempre por mim. Ao Igor, minha força e minha calma, que chegou no meio dessa etapa da minha vida e sempre acreditou em mim. Ao meu orientador, Prof. Dr. José Roberto Trigo, pela oportunidade e pelos muitos ensinamentos. Aos membros da banca e pré-banca, Dr. Alberto Arab, Dra. Daniela Rodrigues, Prof. Dr. Eduardo L. Borba, Prof. Dr. Flavio A. M. dos Santos e Dra. Karina L. Silva- Brandão pelos comentários e sugestões importantes. À FAPESP, pela bolsa concedida. Aos companheiros de laboratório: Kamila, Alberto, Carlos, Marcela, Adriano, Tamara e Aline, pelas discussões, pela ajuda em laboratório, pela amizade e por tornarem o laboratório um ambiente de trabalho divertido. Aos amigos que me ajudaram em campo: Alberto, Ana, Henrique, Thiago e também ao funcionário Paulo Roberto Manzani. Ao funcionário José Carlos da Silva, o Zé, por me ajudar a carregar vasos pesados, quebrar muitos galhos e me ensinar quase tudo o que eu sei sobre cuidados com plantas. À Tamara pela ajuda indispensável com as extrações na etapa final. Às amigas da graduação (Ana, Alê, Bruna, Gaby, Karen, Marilini, Stef e Yara) por estarem comigo nos momentos bons e ruins. Aos muitos amigos do Rio, por entenderem iv minhas longas ausências e sempre me receberem de braços abertos quando eu volto. Aos amigos da pós da Ecologia pelas intensas discussões sobre ecologia ou sobre besteiras, pelos cafés e as cervejinhas. -
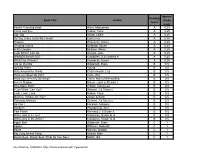
Book Title Author Reading Level Approx. Grade Level
Approx. Reading Book Title Author Grade Level Level Anno's Counting Book Anno, Mitsumasa A 0.25 Count and See Hoban, Tana A 0.25 Dig, Dig Wood, Leslie A 0.25 Do You Want To Be My Friend? Carle, Eric A 0.25 Flowers Hoenecke, Karen A 0.25 Growing Colors McMillan, Bruce A 0.25 In My Garden McLean, Moria A 0.25 Look What I Can Do Aruego, Jose A 0.25 What Do Insects Do? Canizares, S.& Chanko,P A 0.25 What Has Wheels? Hoenecke, Karen A 0.25 Cat on the Mat Wildsmith, Brain B 0.5 Getting There Young B 0.5 Hats Around the World Charlesworth, Liza B 0.5 Have you Seen My Cat? Carle, Eric B 0.5 Have you seen my Duckling? Tafuri, Nancy/Greenwillow B 0.5 Here's Skipper Salem, Llynn & Stewart,J B 0.5 How Many Fish? Cohen, Caron Lee B 0.5 I Can Write, Can You? Stewart, J & Salem,L B 0.5 Look, Look, Look Hoban, Tana B 0.5 Mommy, Where are You? Ziefert & Boon B 0.5 Runaway Monkey Stewart, J & Salem,L B 0.5 So Can I Facklam, Margery B 0.5 Sunburn Prokopchak, Ann B 0.5 Two Points Kennedy,J. & Eaton,A B 0.5 Who Lives in a Tree? Canizares, Susan et al B 0.5 Who Lives in the Arctic? Canizares, Susan et al B 0.5 Apple Bird Wildsmith, Brain C 1 Apples Williams, Deborah C 1 Bears Kalman, Bobbie C 1 Big Long Animal Song Artwell, Mike C 1 Brown Bear, Brown Bear What Do You See? Martin, Bill C 1 Found online, 7/20/2012, http://home.comcast.net/~ngiansante/ Approx.