Inflammation and Endometrial Cancer: a Hypothesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polycystic Ovary Syndrome.Pdf
Female reproductive system diseases Polycystic ovary syndrome Introduction: Polycystic ovary syndrome is one of the most common hormonal disorders among women. The name of this condition comes from the cysts that develop when ovulation vesicles get trapped under the surface of the ovaries preventing them from releasing the eggs. Cause: Normally, the pituitary gland in the brain secretes hormones (FSH) and (LH), that are responsible for controlling ovulation, while the ovary secretes estrogen and progesterone hormones, which prepare the uterus for the egg. The ovary also secretes the male hormone (androgen). However, in the case of polycystic ovary syndrome, the pituitary gland secretes excess amounts of (LH) and the ovary secretes excess amounts of the male hormone (androgen), resulting in irregular menstrual cycles and difficulties conceiving, as well as an increase facial hair and acne. There are many factors that may play a role in causing polycystic ovary syndrome: • Increased resistance to insulin (high blood glucose levels). • Heredity Symptoms: • Menstrual cycle abnormalities: The duration of the menstrual cycle may be prolonged to 35 days, or it could become less frequent occurring less than 8 times a year or it could be completely absent. • Increased body and facial hair • Acne • Obesity • Difficulty conceiving Diagnosis: • Medical history: Absence of the menstrual cycle - increased facial and body hair - acne - excess weight. • Ultrasound examination: of the uterus and ovaries • Blood test: To measure hormone levels, especially androgens and (LH). Treatment: Treatment depends on the symptoms regardless of whether the woman wants to conceive or not: • Lifestyle changes: This includes following a low-carb diet that is rich in grains, vegetables, fruits and small amounts of meat. -

The Discovery of Different Types of Cervical Mucus and the Billings Ovulation Method
The Discovery of Different Types of Cervical Mucus and the Billings Ovulation Method Erik Odeblad Emeritus Professor, Dept. of Medical Biophysics, University of Umeå, Sweden Published with permission from the Bulletin of the Ovulation Method Research and Reference Centre of Australia, 27 Alexandra Parade, North Fitzroy, Victoria 3068, Australia, Volume 21, Number 3, pages 3-35, September 1994. Copyright © Ovulation Method Research and Reference Centre of Australia 1. Abstract 2. Introduction 3. Anatomy and Physiology 4. What is Mucus? 5. The Commencement of my Research 6. The Existence of Different Types of Crypts and of Mucus 7. Identification and Description of G, L, and S Mucus 8. G- and G+ Mucus 9. Age, Pregnancy, the Pill and Microsurgery 10. P Mucus 11. F Mucus 12. The Role of the Vagina 13. The Different Types of Secretions and the Billings Ovulation Method 14. Early Infertile Days 15. The Days of Possible Fertility 16. Late Infertile Days 17. Anovulatory Cycles 18. Lactation 19. Diseases and the Billings Ovulation Method 20. The Future 21. Acknowledgements 22. Author's Note 23. References 24. Appendix Abstract An introduction to and some new anatomical and physiological aspects of the cervix and vagina are presented and also an explanation of the biosynthesis and molecular structure of mucus. The history of my discoveries of the different types of cervical mucus is given. In considering my microbiological investigations I suspected the existence of different types of crypts and cervical mucus and in 1959 1 proved the existence of these different types. The method of examining viscosity by nuclear magnetic resonance was applied to microsamples of mucus extracted 1 outside of several crypts. -

Women's Menstrual Cycles
1 Women’s Menstrual Cycles About once each month during her reproductive years, a woman has a few days when a bloody fluid leaves her womb and passes through her vagina and out of her body. This normal monthly bleeding is called menstruation, or a menstrual period. Because the same pattern happens each month, it is called the menstrual cycle. Most women bleed every 28 days. But some bleed as often as every 20 days or as seldom as every 45 days. Uterus (womb) A woman’s ovaries release an egg once a month. If it is Ovary fertilized she may become pregnant. If not, her monthly bleeding will happen. Vagina Menstruation is a normal part of women’s lives. Knowing how the menstrual cycle affects the body and the ways menstruation changes over a woman’s lifetime can let you know when you are pregnant, and help you detect and prevent health problems. Also, many family planning methods work best when women and men know more about the menstrual cycle (see Family Planning). 17 December 2015 NEW WHERE THERE IS NO DOCTOR: ADVANCE CHAPTERS 2 CHAPTER 24: WOMEN’S MENSTRUAL CYCLES Hormones and the menstrual cycle In women, the hormones estrogen and progesterone are produced mostly in the ovaries, and the amount of each one changes throughout the monthly cycle. During the first half of the cycle, the ovaries make mostly estrogen, which causes the lining of the womb to thicken with blood and tissue. The body makes the lining so a baby would have a soft nest to grow in if the woman became pregnant that month. -

Menstrual Health Glossary Key Words and Acronyms in the Field, from UNICEF + WHO
Menstrual Health Glossary Key words and acronyms in the field, from UNICEF + WHO Menstruation or menses is the natural bodily process of releasing blood and associated matter from the uterus through the vagina as part of the menstrual cycle.¹ A menstruator is a person who menstruates and therefore has menstrual health and hygiene needs – including girls, women, transgender and non-binary persons.¹ Menarche is the onset of menstruation, the time when a girl has her first menstrual period.¹ Menstrual hygiene materials are the products used to catch menstrual flow, such as pads, cloths, tampons or cups. These may also be referred to as menstrual materials or period products.¹ Menstrual Health Glossary Key words and acronyms in the field, from UNICEF + WHO Menstrual supplies are other supportive items needed for MHH, such as body and laundry soap, underwear and pain relief items.¹ Menstrual Hygiene Management (MHM) refers to management of hygiene associated with the menstrual process.¹ Adequate MHM involves: Knowledge and awareness about the menstrual process. Menstrual hygiene materials such as washable pads, disposable pads, tampons, and cups,WASH infrastructure such as Safe, clean, convenient, and private spaces for changing, washing, and/or disposing of menstrual hygiene materials. Adequate amounts of clean water and soap. Supportive social environments that enable menstruators to manage their periods with dignity and confidence. Policies and systems that create positive norms and dismantle limitations associated with menstruation.² Menstrual -
Understanding Your Menstrual Cycle If You're Trying to Conceive
IS MY PERIOD NORMAL? Understanding Your Menstrual Cycle If You’re Trying to Conceive More than 70% 11% 95% of women have or more of of U.S. women start irregular menstrual American women their periods by cycles as menopause suffer from age 16. approaches. endometriosis.1 10% 12% of U.S. women are of women have affected by PCOS trouble getting or (polycystic ovary staying pregnant.3 syndrome).2 Fortunately, your menstrual cycle can tell you a lot about your fertility if you know what to look for. TYPES OF MENSTRUAL CYCLES Only 15% of About Normal = women have 30% of women are fertile only during 21 to 35 days the “perfect” the “normal” fertility 28-day cycle. window—between days 10 and 17 of the menstrual cycle. Day 1 Period starts (aka menses) 27 28 1 2 26 3 25 4 24 5 Day 15-28 23 6 Day 2-14 Luteal phase; Follicular phase; progesterone** 22 WHAT’S NORMAL? 7 FSH released, (follicle- uterine lining 21 8 stimulating matures Give or take a few days, hormone) and a normal cycle looks like this: estrogen released, 20 9 ovulation* begins 19 10 18 11 17 12 16 15 14 13 *ovulation: the process of an ovum (egg) being released from the ovary; occurs 10-14 days before menses. **progesterone: a steroid hormone that tells the uterus to prepare for pregnancy At least 30% of women have an “irregular” cycle either short, long or inconsistent. Short = Long = < 21 days > 35 days May be a sign of: May be a sign of: Hormonal imbalance Hormonal imbalance Ovaries with fewer eggs Lack of ovulation Approach of menopause Other fertility issues Reduced fertility4 Increased risk of miscarriage SIGNS TO WATCH FOR Your menstrual cycle provides valuable clues about your body’s reproductive health. -

A Fixed Formula to Define the Fertile Window of the Menstrual Cycle As the Basis of a Simple Method of Natural Family Planning
ORIGINAL RESEARCH ARTICLE A Fixed Formula to Define the Fertile Window of the Menstrual Cycle as the Basis of a Simple Method of Natural Family Planning Marcos Are´valo,* Irit Sinai,* and Victoria Jennings* A significant number of women worldwide use periodic basis of the proposed Standard Days method, a simple abstinence as their method of family planning. Many of method of natural family planning (NFP). Survey data them use some type of calendar-based approach to deter- from a number of countries around the world show mine when they should abstain from unprotected inter- that a substantial number of women worldwide use course to avoid pregnancy; yet they often lack correct periodic abstinence as their method of family plan- knowledge of when during their menstrual cycle they are ning.1 Many of these women use calendar-based ap- most likely to become pregnant. A simple method of proaches to determine when they should abstain from natural family planning (NFP) based on a fixed formula to unprotected intercourse to avoid pregnancy. How- define the fertile window could be useful to these women. ever, research also indicates that a significant per- This article reports the results of an analysis of the appli- centage of women who claim to use periodic absti- cation of a fixed formula to define the fertile window. A nence lack correct knowledge of when during their large existing data set from a World Health Organization menstrual cycle they are most likely to become study of the Ovulation Method was used to estimate the pregnant.a Most of these women simply abstain from theoretical probability of pregnancy using this formula. -

Natural Family Planning Fact Sheet
Natural Family Planning Fact Sheet ____________________________________________________________________________ 24-hour Emergency Number/Location WHAT’S INSIDE: SOURCES: What is the natural family planning? Office on Women’s Health Basal body temperature method Calendar Method Birth Control Methods: Frequently Cervical Mucus Method Asked Questions How effective are natural family Fertility Awareness planning methods? Advantages of natural family Centers for Disease Control and planning Prevention Drawbacks of natural family planning Unintended Pregnancy Prevention: Contraception U.S. Department of Health & Human Services 200 Independence Avenue, S.W. Washington, D.C. Oklahoma State Department of Health ODH Form 337 MCH/Perinatal & Reproductive Health Division/Family Planning Program Revised Oct 2014 Office of Population Affairs Natural Family Planning Fact Sheet How effective is natural family planning? Of 100 couples who use natural family planning methods each year, anywhere from 1 to 25 will become pregnant. Natural family planning can be an effective type of birth control if all three methods are used and if all are always used correctly. What is natural family planning? A woman with a normal menstrual cycle has about 8 days a month when she can get pregnant. These include the five days before she ovulates (when an egg is released), the day she ovulates, and about one to two days after ovulation. Natural family planning (sometimes known as fertility awareness or the rhythm method) is an approach to birth control some couples use to predict when these fertile days happen. It involves paying close attention to the menstrual cycle by using methods that include: Basal Body Temperature Method Calendar Method Cervical Mucus Method When all three methods are used together, it is known as the symptothermal method. -

Age and Fertility: a Guide for Patients
Age and Fertility A Guide for Patients PATIENT INFORMATION SERIES Published by the American Society for Reproductive Medicine under the direction of the Patient Education Committee and the Publications Committee. No portion herein may be reproduced in any form without written permission. This booklet is in no way intended to replace, dictate or fully define evaluation and treatment by a qualified physician. It is intended solely as an aid for patients seeking general information on issues in reproductive medicine. Copyright © 2012 by the American Society for Reproductive Medicine AMERICAN SOCIETY FOR REPRODUCTIVE MEDICINE Age and Fertility A Guide for Patients Revised 2012 A glossary of italicized words is located at the end of this booklet. INTRODUCTION Fertility changes with age. Both males and females become fertile in their teens following puberty. For girls, the beginning of their reproductive years is marked by the onset of ovulation and menstruation. It is commonly understood that after menopause women are no longer able to become pregnant. Generally, reproductive potential decreases as women get older, and fertility can be expected to end 5 to 10 years before menopause. In today’s society, age-related infertility is becoming more common because, for a variety of reasons, many women wait until their 30s to begin their families. Even though women today are healthier and taking better care of themselves than ever before, improved health in later life does not offset the natural age-related decline in fertility. It is important to understand that fertility declines as a woman ages due to the normal age- related decrease in the number of eggs that remain in her ovaries. -

Endometrial Hyperplasia
AQ The American College of Obstetricians and Gynecologists FREQUENTLY ASKED QUESTIONS FAQ147 GYNECOLOGIC PROBLEMS Endometrial Hyperplasia • What is endometrial hyperplasia? • How does the endometrium normally change throughoutf the menstrual cycle? • What causes endometrial hyperplasia? • When does endometrial hyperplasia occur? • What risk factors are associated with endometrial hyperplasia? • What are the types of endometrial hyperplasia? • What are signs and symptoms of endometrial hyperplasia? • How is endometrial hyperplasia diagnosed? • What treatments are available for endometrial hyperplasia? • What can I do to help prevent endometrial hyperplasia? • Glossary What is endometrial hyperplasia? Endometrial hyperplasia occurs when the endometrium, the lining of the uterus, becomes too thick. It is not cancer, but in some cases, it can lead to cancer of the uterus. How does the endometrium normally change throughout the menstrual cycle? The endometrium changes throughout the menstrual cycle in response to hormones. During the first part of the cycle, the hormone estrogen is made by the ovaries. Estrogen causes the lining to grow and thicken to prepare the uterus for pregnancy. In the middle of the cycle, an egg is released from one of the ovaries (ovulation). Following ovulation, levels of another hormone called progesterone begin to increase. Progesterone prepares the endometrium to receive and nourish a fertilized egg. If pregnancy does not occur, estrogen and progesterone levels decrease. The decrease in progesterone triggers menstruation, or shedding of the lining. Once the lining is completely shed, a new menstrual cycle begins. What causes endometrial hyperplasia? Endometrial hyperplasia most often is caused by excess estrogen without progesterone. If ovulation does not occur, progesterone is not made, and the lining is not shed. -

Menstrual Cycle Patterns and Their Determinants During the Menopausal Transition Among a Multiethnic Cohort of Women
Menstrual Cycle Patterns and Their Determinants during the Menopausal Transition among a Multiethnic Cohort of Women by Pangaja Paramsothy A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Epidemiological Science) in The University of Michigan 2012 Doctoral Committee: Professor Siobán D. Harlow, Chair Professor John F. Randolph Jr. Associate Professor Michael R. Elliott Associate Professor Lynda D. Lisabeth © Pangaja Paramsothy 2012 ACKNOWLEDGEMENTS I first like to thank my academic advisor and dissertation committee chair, Dr. Harlow for her mentorship, encouragement, and support during my time here at the University of Michigan. I would also like to thank my dissertation committee: Dr. Randolph, Dr. Elliott, and Dr. Lisabeth for their support, guidance, and feedback. Their involvement strengthened this work. I would like to thank Matheos Yosef for sharing his statistical expertise with quantile regression. His patience and kindness is very much appreciated. I would also like to thank Nancy Vander Kuyl for her administrative support and most importantly her moral support and her cheerleading throughout my time here. I would like to thank my SWAN site coauthors: Dr. Crawford, Dr. Gold, and Dr. Greendale for their timely feedback and suggestions. I am indebted to the SWAN staff at all study sites for their years of hard work. I would also like to express my gratitude to the SWAN participants. Their steadfast dedication in filling out the SWAN Menstrual Calendar made this research possible. I gratefully acknowledge funding support from the Rackham Graduate School of University of Michigan as well as the Department of Epidemiology, University of Michigan School of Public Health. -

TIP 51 Substance Abuse Treatment Addressing the Specific Needs Of
Substance Abuse Treatment: Addressing the Specific Needs of Women A Treatment Improvement Protocol TIP 51 Substance Abuse Treatment: Addressing the Specific Needs of Women A Treatment Improvement Protocol TIP 51 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Substance Abuse and Mental Health Services Administration 1 Choke Cherry Road Rockville, MD 20857 Acknowledgments Recommended Citation This publication was prepared under contract Substance Abuse and Mental Health Services numbers 270-99-7072 and 270-04-7049 by the Administration. Substance Abuse Treatment: Knowledge Application Program (KAP), a Joint Addressing the Specific Needs of Women. Treat Venture of The CDM Group, Inc., and JBS ment Improvement Protocol (TIP) Series, No. International, Inc., for the Substance Abuse 51. HHS Publication No. (SMA) 13-4426. Rock and Mental Health Services Administration ville, MD: Substance Abuse and Mental Health (SAMHSA), U.S. Department of Health and Hu Services Administration, 2009. man Services (HHS). Andrea Kopstein, Ph.D., M.P.H., Karl D. White, Ed.D., and Christina Currier served as the Contracting Officer’s Rep Originating Office resentatives. Quality Improvement and Workforce Develop ment Branch, Division of Services Improve Disclaimer ment, Center for Substance Abuse Treatment, Substance Abuse and Mental Health Services The views, opinions, and content of this publi Administration, 1 Choke Cherry Road, Rock cation are those of the author and do not neces ville, MD 20857. sarily reflect the views, opinions, or policies of SAMHSA or HHS. HHS Publication No. (SMA) 13-4426 Public Domain Notice First Printed 2009 All material appearing in this report is in the Revised 2010, 2012, and 2013 public domain and may be reproduced or copied without permission from SAMHSA. -
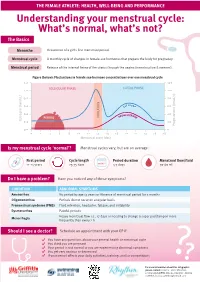
Understanding Your Menstrual Cycle: What's Normal, What's Not?
THE FEMALE ATHLETE: HEALTH, WELL-BEING AND PERFORMANCE Understanding your menstrual cycle: What’s normal, what’s not? The Basics Menarche Occurrence of a girl’s first menstrual period. Menstrual cycle A monthly cycle of changes in female-sex hormones that prepare the body for pregnancy. Menstrual period Release of the internal lining of the uterus through the vagina (menstruation & menses). Figure (below): Fluctuations in female sex-hormone concentrations over one menstrual cycle 1.2 120 FOLLICULAR PHASE LUTEAL PHASE 1.0 100 0.8 80 0.6 60 0.4 40 PERIOD 0.2 20 0.0 0 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 Is my menstrual cycle ‘normal’? Menstrual cycles vary, but are on average: First period Cycle length Period duration Menstrual flow/fluid 11-14 years 25-35 days 4-7 days 30-60 mL Do I have a problem? Have you noticed any of these symptoms? CONDITION ABNORMAL SYMPTOMS Amenorrhea No period by age 15 years or Absence of menstrual period for 3 months Oligomenorrhea Periods do not occur on a regular basis Premenstrual syndrome (PMS) Fluid retention, headache, fatigue, and irritability Dysmenorrhea Painful periods Menorrhagia Heavy menstrual flow i.e., >7 days or needing to change a super pad/tampon more frequently than every 2 h Should I see a doctor? Schedule an appointment with your GP if: You have any questions about your general health or menstrual cycle You think you are pregnant Your period is not normal or you are experiencing abnormal symptoms You get very anxious or depressed If your period affects your daily activities, training, and/or competitions ?? For more information about this infographic, please contact: Content - Clare Minahan, [email protected], Designer - Bianca Cattelini, [email protected].