UC Irvine UC Irvine Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supp Material.Pdf
Legends for Supplemental Figures and Tables Figure S1. Expression of Tlx during retinogenesis. (A) Staged embryos were stained for β- galactosidase knocked into the Tlx locus to indicate Tlx expression. Tlx was expressed in the neural blast layer in the early phase of neural retina development (blue signal). (B) Expression of Tlx in neural retina was quantified using Q-PCR at multiple developmental stages. Figure S2. Expression of p27kip1 and cyclin D1 (Ccnd1) at various developmental stages in wild-type or Tlx-/- retinas. (A) Q-PCR analysis of p27kip1 mRNA expression. (B) Western blotting analysis of p27kip1 protein expression. (C) Q-PCR analysis of cyclin D1 mRNA expression. Figure S3. Q-PCR analysis of mRNA expression of Sf1 (A), Lrh1 (B), and Atn1 (C) in wild-type mouse retinas. RNAs from testis and liver were used as controls. Table S1. List of genes dysregulated both at E15.5 and P0 Tlx-/- retinas. Gene E15.5 P0 Cluste Gene Title Fold Fold r Name p-value p-value Change Change nuclear receptor subfamily 0, group B, Nr0b1 1.65 0.0024 2.99 0.0035 member 1 1 Pou4f3 1.91 0.0162 2.39 0.0031 POU domain, class 4, transcription factor 3 1 Tcfap2d 2.18 0.0000 2.37 0.0001 transcription factor AP-2, delta 1 Zic5 1.66 0.0002 2.02 0.0218 zinc finger protein of the cerebellum 5 1 Zfpm1 1.85 0.0030 1.88 0.0025 zinc finger protein, multitype 1 1 Pten 1.60 0.0155 1.82 0.0131 phospatase and tensin homolog 2 Itgb5 -1.85 0.0063 -1.85 0.0007 integrin beta 5 2 Gpr49 6.86 0.0001 15.16 0.0001 G protein-coupled receptor 49 3 Cmkor1 2.60 0.0007 2.72 0.0013 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Prox1regulates the Subtype-Specific Development of Caudal Ganglionic
The Journal of Neuroscience, September 16, 2015 • 35(37):12869–12889 • 12869 Development/Plasticity/Repair Prox1 Regulates the Subtype-Specific Development of Caudal Ganglionic Eminence-Derived GABAergic Cortical Interneurons X Goichi Miyoshi,1 Allison Young,1 Timothy Petros,1 Theofanis Karayannis,1 Melissa McKenzie Chang,1 Alfonso Lavado,2 Tomohiko Iwano,3 Miho Nakajima,4 Hiroki Taniguchi,5 Z. Josh Huang,5 XNathaniel Heintz,4 Guillermo Oliver,2 Fumio Matsuzaki,3 Robert P. Machold,1 and Gord Fishell1 1Department of Neuroscience and Physiology, NYU Neuroscience Institute, Smilow Research Center, New York University School of Medicine, New York, New York 10016, 2Department of Genetics & Tumor Cell Biology, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, 3Laboratory for Cell Asymmetry, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan, 4Laboratory of Molecular Biology, Howard Hughes Medical Institute, GENSAT Project, The Rockefeller University, New York, New York 10065, and 5Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724 Neurogliaform (RELNϩ) and bipolar (VIPϩ) GABAergic interneurons of the mammalian cerebral cortex provide critical inhibition locally within the superficial layers. While these subtypes are known to originate from the embryonic caudal ganglionic eminence (CGE), the specific genetic programs that direct their positioning, maturation, and integration into the cortical network have not been eluci- dated. Here, we report that in mice expression of the transcription factor Prox1 is selectively maintained in postmitotic CGE-derived cortical interneuron precursors and that loss of Prox1 impairs the integration of these cells into superficial layers. Moreover, Prox1 differentially regulates the postnatal maturation of each specific subtype originating from the CGE (RELN, Calb2/VIP, and VIP). -

Systematic Detection of Divergent Brain Proteins in Human Evolution and Their Roles in Cognition
bioRxiv preprint doi: https://doi.org/10.1101/658658; this version posted June 3, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Systematic detection of divergent brain proteins in human evolution and their roles in cognition Guillaume Dumas1,*, Simon Malesys1 and Thomas Bourgeron1 Affiliations: 1 Human Genetics and Cognitive Functions, Institut Pasteur, UMR3571 CNRS, Université de Paris, Paris, (75015) France * Corresponding author: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/658658; this version posted June 3, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract The human brain differs from that of other primates, but the underlying genetic mechanisms remain unclear. Here we measured the evolutionary pressures acting on all human protein- coding genes (N=17,808) based on their divergence from early hominins such as Neanderthal, and non-human primates. We confirm that genes encoding brain-related proteins are among the most conserved of the human proteome. Conversely, several of the most divergent proteins in humans compared to other primates are associated with brain-associated diseases such as micro/macrocephaly, dyslexia, and autism. We identified specific eXpression profiles of a set of divergent genes in ciliated cells of the cerebellum, that might have contributed to the emergence of fine motor skills and social cognition in humans. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

2020 Program Book
PROGRAM BOOK Note that TAGC was cancelled and held online with a different schedule and program. This document serves as a record of the original program designed for the in-person meeting. April 22–26, 2020 Gaylord National Resort & Convention Center Metro Washington, DC TABLE OF CONTENTS About the GSA ........................................................................................................................................................ 3 Conference Organizers ...........................................................................................................................................4 General Information ...............................................................................................................................................7 Mobile App ....................................................................................................................................................7 Registration, Badges, and Pre-ordered T-shirts .............................................................................................7 Oral Presenters: Speaker Ready Room - Camellia 4.......................................................................................7 Poster Sessions and Exhibits - Prince George’s Exhibition Hall ......................................................................7 GSA Central - Booth 520 ................................................................................................................................8 Internet Access ..............................................................................................................................................8 -

Co-Occurrence Based Meta-Analysis of Scientific Texts
Vol. 21 no. 9 2005, pages 2049–2058 BIOINFORMATICS ORIGINAL PAPER doi:10.1093/bioinformatics/bti268 Data and text mining Co-occurrence based meta-analysis of scientific texts: retrieving biological relationships between genes R. Jelier1,∗ G. Jenster2, L. C. J. Dorssers3, C. C. van der Eijk1, E. M. van Mulligen1, B. Mons1 and J. A. Kors1 1Department of Medical Informatics, 2Department of Urology and 3Department of Pathology, Erasmus MC—University Medical Center, Rotterdam, The Netherlands Received on July 15, 2004; revised on December 22, 2004; accepted on January 6, 2005 Advance Access publication January 18, 2005 ABSTRACT are studied, the number of relevant publications will frequently be Motivation: The advent of high-throughput experiments in molecu- prohibitively large. This renders the traditional approach of manu- lar biology creates a need for methods to efficiently extract and use ally searching bibliographic databases for every gene and reading information for large numbers of genes. Recently, the associative scientific articles inadequate. It is therefore an important challenge concept space (ACS) has been developed for the representation of at this time to make the available information both accessible and information extracted from biomedical literature. The ACS is a Euc- interpretable for molecular biologists. lidean space in which thesaurus concepts are positioned and the An interesting current development is the use of annotations of distances between concepts indicates their relatedness. The ACS genes with gene ontology (GO) terms (Ashburner et al., 2000; Camon uses co-occurrence of concepts as a source of information. In this et al., 2004) for the analysis of the results of microarray experiments paper we evaluate how well the system can retrieve functionally (Zhang et al., 2004; Al Shahrour et al., 2004). -

Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation
Molecular Cell Article Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation Yanzhong Yang,1 Kevin M. McBride,1 Sean Hensley,1 Yue Lu,1 Frederic Chedin,2 and Mark T. Bedford1,* 1The University of Texas MD Anderson Cancer Center, P.O. Box 389, Smithville, TX 78957, USA 2Department of Molecular & Cellular Biology, The University of California at Davis, Davis, CA 95616, USA *Correspondence: [email protected] http://dx.doi.org/10.1016/j.molcel.2014.01.011 SUMMARY nine methyltransferase 1), which deposit the H4R3me2a and H3R17me2a marks, respectively (Yang and Bedford, 2013). Tudor domain-containing protein 3 (TDRD3) is a Both of these marks are recognized by the Tudor domain of major methylarginine effector molecule that reads TDRD3 (Yang et al., 2010), a protein that is enriched at the pro- methyl-histone marks and facilitates gene transcrip- moters of highly transcribed genes and can likely also associate tion. However, the underlying mechanism by which with the C-terminal domain (CTD) of RNA Polymerase II (RNAP II) TDRD3 functions as a transcriptional coactivator is (Sims et al., 2011). TDRD3 has no enzymatic activity of its unknown. We identified topoisomerase IIIB (TOP3B) own, but here we show that it is tightly complexed with DNA topoisomerase IIIb (TOP3B), an interaction that bestows, in as a component of the TDRD3 complex. TDRD3 part, coactivator activity on TDRD3. TOP3B is a member of serves as a molecular bridge between TOP3B and the 1A subfamily of DNA topoisomerases and, as such, targets arginine-methylated histones. The TDRD3-TOP3B underwound or negatively supercoiled DNA (Wang, 2002). -

MTHFR) and Methionine Synthase Reductase (MTRR
ANTICANCER RESEARCH 28 : 2807-2812 (2008) Methylenetetrahy drofolate Reductase (MTHFR) and Methionine Synthase Reductase (MTRR) Gene Polymorphisms as Risk Factors for Hepatocellular Carcinoma in a Korean Population SUN YOUNG KWAK 1,2 , UN KYUNG KIM 3, HYO JIN CHO 2, HEE KEUN LEE 3, HYE JIN KIM 2, NAM KEUN KIM 2 and SEONG GYU HWANG 1,2 1Department of Internal Medicine, 2Institute for Clinical Research, College of Medicine, Pochon CHA University, Seongnam; 3Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, South Korea Abstract. Hepatocellular carcinoma (HCC) is the third were increased risk factors for the disease. The MTHFR most frequent cause of cancer death in South Korea, but 1298A>C and the MTRR 66A>G genotypes were associated genetic susceptibility factors of HCC have not been examined with an increased risk of HCC in th is Korean population. extensively. Methylenetetrahydrofolate reductase (MTHFR) Further studies involving larger and varied populations and methionine synthase reductase (MTRR) play an essential could provide a potential tool for cancer risk assessment in role in both DNA synthesis and methylation and patients who are at risk of developing HCC. polymorphisms in the MTHFR gene, 677C>T, 1298A>C and the MTRR gene, 66A>G, are associated with several types of Hepatocellular carcinoma (HCC) is one of the most common malignancy. In this study, the allelic frequencies and malignant tumors with the third highest mortality rate among genotype distribution of three polymorphisms in the MTHFR malignancies in South Korea. According to a statistical report and MTRR genes from 96 hepatocellular carcinoma (HCC) in 2006, it occurred in 33.8/10,000 men and 11.2/10,000 patients and 201 controls were examined to assess the women in South Korea in 2005 (1). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
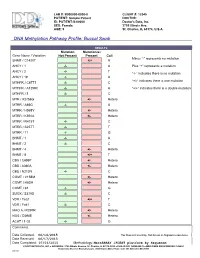
DNA Methylation Pathway Profile; Buccal Swab
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-00000 Doctor's Data, Inc. SEX: Female 3755 Illinois Ave. AGE: 5 St. Charles, IL 60174, U.S.A. !DNA Methylation Pathway Profile; Buccal Swab RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call Minus “-“ represents no mutation SHMT / C1420T +/+ A AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A “+/-“ indicates there is one mutation MTHFR / C677T -/- C MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G -/- A MTRR / H595Y +/- Hetero MTRR / K350A +/- Hetero MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 -/- G BHMT / 1 -/- A BHMT / 2 -/- C BHMT / 4 +/- Hetero BHMT / 8 +/+ T CBS / C699T +/- Hetero CBS / A360A +/- Hetero CBS / N212N -/- C COMT / V158M +/- Hetero COMT / H62H +/- Hetero COMT / 61 -/- G SUOX / S370S -/- C VDR / Taq1 +/+ T VDR / Fok1 -/- C MAO A / R297R +/- Hetero NOS / D298E +/- Hetero ACAT / 1-02 -/- G Comments: Date Collected: 06/14/2015 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 06/17/2015 Date Completed: 07/03/2015 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare.