Narrowing the Genetic Causes of Language Dysfunction in the 1Q21.1 Microduplication Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Mutational Landscape of Patients with Chronic Lymphocytic Leukemia
Analysis of mutational landscape of patients with chronic lymphocytic leukemia A.V. Terskikh1, A.A. Samsonova2, A.A. Kanapin2 1-Peter the Great St.Petersburg Polytechnic University, St. Petersburg, Russia, anastasiya- [email protected]; 2-Department of Oncology, University of Oxford, Oxford, UK, [email protected], [email protected] A precise understanding of the genomic features of chronic lymphocytic leukemia (CLL) may benefit the study of the disease’s staging and treatment. Genomic landscape of CLL probably reflects either an unknown underlying biochemical mechanism playing a key role in CLL or multiple biochemical pathways independently driving the development of this tumor. The elucidation of either scenario may have important consequences on the clinical management of CLL. Our aim is to analyze mutational landscape of the disease to identify potential pathways driving the pathology. Chronic lymphocytic leukemia is a clonal neoplasia of B-lymphocytes which accumulate mainly in the blood, bone marrow, lymph nodes and spleen [1]. Notably, these B- lymphocytes are differentiated, and can remain in an arrested state for several years after diagnosis. Two major molecular subtypes can be distinguished, characterized respectively by a high or low number of somatic hypermutations in the variable region of immunoglobulin genes [2]. This classification system was improved with the characterization of additional genomic and transcriptomic factors [3]. However, the genomic events that dictate the initiation and heterogeneous evolution of CLL remained partially unknown. Next-Generation Sequencing (NGS) allows the comparison of the genome of tumor cells with the constitutive genome in normal tissues of the same patients. The variants present in the tumor genome and absent from the germinal genome are called somatic mutations, and constitute a requisite of cancer development. -

Novel Genetic Features of Human and Mouse Purkinje Cell Differentiation Defined by Comparative Transcriptomics
Novel genetic features of human and mouse Purkinje cell differentiation defined by comparative transcriptomics David E. Buchholza, Thomas S. Carrollb, Arif Kocabasa, Xiaodong Zhua, Hourinaz Behestia, Phyllis L. Faustc, Lauren Stalbowa, Yin Fanga, and Mary E. Hattena,1 aLaboratory of Developmental Neurobiology, The Rockefeller University, New York, NY 10065; bBioinformatics Resource Center, The Rockefeller University, New York, NY 10065; and cDepartment of Pathology and Cell Biology, Columbia University Irving Medical Center and the New York Presbyterian Hospital, New York, NY 10032 Contributed by Mary E. Hatten, April 22, 2020 (sent for review January 3, 2020; reviewed by Lorenz Studer and Hynek Wichterle) Comparative transcriptomics between differentiating human plu- is ataxia-telangiectasia, which shows massive loss of PCs in hu- ripotent stem cells (hPSCs) and developing mouse neurons offers a mans but not in the mouse (11). Human pluripotent stem cells powerful approach to compare genetic and epigenetic pathways (hPSCs) provide a complementary approach to studying human in human and mouse neurons. To analyze human Purkinje cell disease in the mouse (12–14). Validated methods to derive (PC) differentiation, we optimized a protocol to generate human specific neural subtypes from hPSCs are necessary prerequisites pluripotent stem cell-derived Purkinje cells (hPSC-PCs) that formed to disease modeling. We and others have recently developed synapses when cultured with mouse cerebellar glia and granule protocols to derive PCs from hPSCs (15–18). cells and fired large calcium currents, measured with the geneti- One limitation of most hPSC-derived central nervous system cally encoded calcium indicator jRGECO1a. To directly compare (CNS) neurons is the lack of genetic information, especially of global gene expression of hPSC-PCs with developing mouse PCs, transcriptomic signatures, to rigorously identify specific types of we used translating ribosomal affinity purification (TRAP). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

MAP17's Up-Regulation, a Crosspoint in Cancer and Inflammatory
García-Heredia and Carnero Molecular Cancer (2018) 17:80 https://doi.org/10.1186/s12943-018-0828-7 REVIEW Open Access Dr. Jekyll and Mr. Hyde: MAP17’s up- regulation, a crosspoint in cancer and inflammatory diseases José M. García-Heredia1,2,3 and Amancio Carnero1,3* Abstract Inflammation is a common defensive response that is activated after different harmful stimuli. This chronic, or pathological, inflammation is also one of the causes of neoplastic transformation and cancer development. MAP17 is a small protein localized to membranes with a restricted pattern of expression in adult tissues. However, its expression is common in destabilized cells, as it is overexpressed both in inflammatory diseases and in cancer. MAP17 is overexpressed in most, if not all, carcinomas and in many tumors of mesenchymal origin, and correlates with higher grade and poorly differentiated tumors. This overexpression drives deep changes in cell homeostasis including increased oxidative stress, deregulation of signaling pathways and increased growth rates. Importantly, MAP17 is associated in tumors with inflammatory cells infiltration, not only in cancer but in various inflammatory diseases such as Barret’s esophagus, lupus, Crohn’s, psoriasis and COPD. Furthermore, MAP17 also modifies the expression of genes connected to inflammation, showing a clear induction of the inflammatory profile. Since MAP17 appears highly correlated with the infiltration of inflammatory cells in cancer, is MAP17 overexpression an important cellular event connecting tumorigenesis and inflammation? Keywords: MAP17, Cancer, Inflammatory diseases Background process ends when the activated cells undergo apoptosis in Inflammatory response is a common defensive process acti- a highly regulated process that finishes after pathogens and vated after different harmful stimuli, constituting a highly cell debris have been phagocytized [9]. -

Paired Involvement of Human-Specific Olduvai Domains and NOTCH2NL
Human Genetics (2019) 138:715–721 https://doi.org/10.1007/s00439-019-02018-4 ORIGINAL INVESTIGATION Paired involvement of human‑specifc Olduvai domains and NOTCH2NL genes in human brain evolution Ian T. Fiddes1 · Alex A. Pollen2 · Jonathan M. Davis3 · James M. Sikela3 Received: 31 October 2018 / Accepted: 16 April 2019 / Published online: 13 May 2019 © The Author(s) 2019 Abstract Sequences encoding Olduvai (DUF1220) protein domains show the largest human-specifc increase in copy number of any coding region in the genome and have been linked to human brain evolution. Most human-specifc copies of Olduvai (119/165) are encoded by three NBPF genes that are adjacent to three human-specifc NOTCH2NL genes that have been shown to promote cortical neurogenesis. Here, employing genomic, phylogenetic, and transcriptomic evidence, we show that these NOTCH2NL/NBPF gene pairs evolved jointly, as two-gene units, very recently in human evolution, and are likely co-regulated. Remarkably, while three NOTCH2NL paralogs were added, adjacent Olduvai sequences hyper-amplifed, adding 119 human- specifc copies. The data suggest that human-specifc Olduvai domains and adjacent NOTCH2NL genes may function in a coordinated, complementary fashion to promote neurogenesis and human brain expansion in a dosage-related manner. Introduction part of these eforts, genomic factors are beginning to be uncovered that potentially contributed to the evolutionary The increasing availability of primate genomic data is pro- expansion of the human brain (Sikela 2006; O’Bleness et al. viding a unique opportunity to identify lineage-specifc 2012a). While there are a number of genomic mechanisms sequence diferences among human and other primates. -

Molecular Distinctions Between Pediatric and Adult Mature B-Cell Non-Hodgkin Lymphomas Identified Through Genomic Profiling
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Public Health Resources Public Health Resources 4-19-2012 Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling Karen Deffenbacher University of Nebraska Medical Center, Omaha Javeed Iqbal University of Nebraska Medical Center, Omaha Warren Sanger University of Nebraska Medical Center, Omaha Yulei Shen University of Nebraska Medical Center, Omaha Cynthia Lachel University of Nebraska Medical Center, Omaha See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/publichealthresources Part of the Public Health Commons Deffenbacher, Karen; Iqbal, Javeed; Sanger, Warren; Shen, Yulei; Lachel, Cynthia; Liu, Zhongfeng; Liu, Yanyan; Lim, Megan; Perkins, Sherrie; Fu, Kai; Smith, Lynette; Lynch, James; Staudt, Louis; Rimsza, Lisa M.; Jaffe, Elaine; Rosenwald, Andreas; Ott, German; Delabie, Jan; Campo, Elias; Gascoyne, Randy; Cairo, Mitchell; Weisenburger, Dennis; Greiner, Timothy; Gross, Thomas; and Chan, Wing, "Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling" (2012). Public Health Resources. 145. https://digitalcommons.unl.edu/publichealthresources/145 This Article is brought to you for free and open access by the Public Health Resources at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Public Health Resources by an authorized administrator -

Evolution and Diversity of Copy Number Variation in the Great Ape Lineage
Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Evolution and diversity of copy number variation in the great ape lineage Peter H. Sudmant1, John Huddleston1,7, Claudia R. Catacchio2, Maika Malig1, LaDeana W. Hillier3, Carl Baker1, Kiana Mohajeri1, Ivanela Kondova4, Ronald E. Bontrop4, Stephan Persengiev4, Francesca Antonacci2, Mario Ventura2, Javier Prado-Martinez5, Tomas 5,6 1,7 Marques-Bonet , and Evan E. Eichler 1. Department of Genome Sciences, University of Washington, Seattle, WA, USA 2. University of Bari, Bari, Italy 3. The Genome Institute, Washington University School of Medicine, St. Louis, MO, USA 4. Department of Comparative Genetics, Biomedical Primate Research Centre, Rijswijk, The Netherlands 5. Institut de Biologia Evolutiva, (UPF-CSIC) Barcelona, Spain 6. Institució Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain 7. Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA Correspondence to: Evan Eichler Department of Genome Sciences University of Washington School of Medicine Foege S-413A, Box 355065 3720 15th Ave NE Seattle, WA 98195 E-mail: [email protected] 1 Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Copy number variation (CNV) contributes to the genetic basis of disease and has significantly restructured the genomes of humans and great apes. The diversity and rate of this process, however, has not been extensively explored among the great ape lineages. We analyzed 97 deeply sequenced great ape and human genomes and estimate that 16% (469 Mbp) of the hominid genome has been affected by recent copy number changes. -
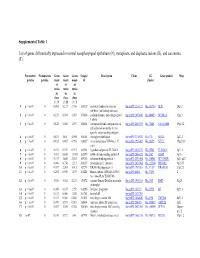
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

The Driver of Extreme Human-Specific Olduvai Repeat Expansion Remains Highly Active in the Human
Genetics: Early Online, published on November 21, 2019 as 10.1534/genetics.119.302782 1 The Driver of Extreme Human-Specific Olduvai Repeat Expansion Remains Highly Active in the Human 2 Genome 3 Ilea E. Heft,1,* Yulia Mostovoy,2,* Michal Levy-Sakin,2 Walfred Ma,2 Aaron J. Stevens,3 Steven 4 Pastor,4 Jennifer McCaffrey,4 Dario Boffelli,5 David I. Martin,5 Ming Xiao,4 Martin A. Kennedy,3 5 Pui-Yan Kwok,2,6,7 and James M. Sikela1 6 7 1Department of Biochemistry and Molecular Genetics, and Human Medical Genetics and 8 Genomics Program, University of Colorado School of Medicine, Aurora CO 80045 9 2Cardiovascular Research Institute, 6Department of Dermatology, and 7Institute for Human 10 Genetics, University of California, San Francisco, San Francisco, CA, USA 11 3Department of Pathology, University of Otago, Christchurch, New Zealand 8140 12 4School of Biomedical Engineering, Drexel University, Philadelphia, PA 19104 13 5Children's Hospital Oakland Research Institute, Oakland, CA, 94609 14 15 Corresponding author: James M. Sikela: [email protected] 16 *Ilea E. Heft and Yulia Mostovoy contributed equally to this article. 17 1 Copyright 2019. 18 Abstract 19 Sequences encoding Olduvai protein domains (formerly DUF1220) show the greatest 20 human lineage-specific increase in copy number of any coding region in the genome and have 21 been associated, in a dosage-dependent manner, with brain size, cognitive aptitude, autism, 22 and schizophrenia. Tandem intragenic duplications of a three-domain block, termed the Olduvai 23 triplet, in four NBPF genes in the chromosomal 1q21.1-.2 region are primarily responsible for 24 the striking human-specific copy number increase. -

Prenatal Detection of TAR Syndrome in a Fetus with Compound Inheritance of an RBM8A SNP and a 334‑Kb Deletion: a Case Report
MOLECULAR MEDICINE REPORTS 9: 163-165, 2014 Prenatal detection of TAR syndrome in a fetus with compound inheritance of an RBM8A SNP and a 334‑kb deletion: A case report IOANNIS PAPOULIDIS1, EIRINI OIKONOMIDOU1, SANDRO ORRU2, ELISAVET SIOMOU1, MARIA KONTODIOU1, MAKARIOS ELEFTHERIADES3, VASILIOS BACOULAS4, JUAN C. CIGUDOSA5, JAVIER SUELA5, LORETTA THOMAIDIS6 and EMMANOUIL MANOLAKOS1,2 1Laboratory of Genetics, Eurogenetica S.A., Thessaloniki 55133, Greece; 2Department of Medical Genetics, University of Cagliari, Binaghi Hospital, Cagliari I‑09126, Italy; 3Embryocare, Fetal Medicine Unit, Athens 11522; 4Fetal Medicine Centre, Athens 10674, Greece; 5NIMGenetics, Madrid 28049, Spain; 6Department of Pediatrics, Aglaia Kyriakou Children's Hospital, University of Athens, Athens 11527, Greece Received May 30, 2013; Accepted October 16, 2013 DOI: 10.3892/mmr.2013.1788 Abstract. Thrombocytopenia-absent radius syndrome identified the presence of a minimally deleted 200‑kb region (TAR) is a rare genetic disorder that is characterized by the at chromosome band 1q21.1 in patients with TAR, but it is not absence of the radius bone in each forearm and a markedly sufficient to cause the phenotype (3,4). A study identified two reduced platelet count that results in life-threatening bleeding rare single nucleotide polymorphisms (SNPs) in the regulatory episodes (thrombocytopenia). Tar syndrome has been associ- region of the RBM8A gene that are involved in TAR syndrome ated with a deletion of a segment of 1q21.1 cytoband. The through the reduction of the expression of the RBM8A-encoded 1q21.1 deletion syndrome phenotype includes Tar and other Y14 protein (4). The first allele (rs139428292 G>A), which is features such as mental retardation, autism and microcephaly. -

Supplementary Table S1. List of Differentially Expressed
Supplementary table S1. List of differentially expressed transcripts (FDR adjusted p‐value < 0.05 and −1.4 ≤ FC ≥1.4). 1 ID Symbol Entrez Gene Name Adj. p‐Value Log2 FC 214895_s_at ADAM10 ADAM metallopeptidase domain 10 3,11E‐05 −1,400 205997_at ADAM28 ADAM metallopeptidase domain 28 6,57E‐05 −1,400 220606_s_at ADPRM ADP‐ribose/CDP‐alcohol diphosphatase, manganese dependent 6,50E‐06 −1,430 217410_at AGRN agrin 2,34E‐10 1,420 212980_at AHSA2P activator of HSP90 ATPase homolog 2, pseudogene 6,44E‐06 −1,920 219672_at AHSP alpha hemoglobin stabilizing protein 7,27E‐05 2,330 aminoacyl tRNA synthetase complex interacting multifunctional 202541_at AIMP1 4,91E‐06 −1,830 protein 1 210269_s_at AKAP17A A‐kinase anchoring protein 17A 2,64E‐10 −1,560 211560_s_at ALAS2 5ʹ‐aminolevulinate synthase 2 4,28E‐06 3,560 212224_at ALDH1A1 aldehyde dehydrogenase 1 family member A1 8,93E‐04 −1,400 205583_s_at ALG13 ALG13 UDP‐N‐acetylglucosaminyltransferase subunit 9,50E‐07 −1,430 207206_s_at ALOX12 arachidonate 12‐lipoxygenase, 12S type 4,76E‐05 1,630 AMY1C (includes 208498_s_at amylase alpha 1C 3,83E‐05 −1,700 others) 201043_s_at ANP32A acidic nuclear phosphoprotein 32 family member A 5,61E‐09 −1,760 202888_s_at ANPEP alanyl aminopeptidase, membrane 7,40E‐04 −1,600 221013_s_at APOL2 apolipoprotein L2 6,57E‐11 1,600 219094_at ARMC8 armadillo repeat containing 8 3,47E‐08 −1,710 207798_s_at ATXN2L ataxin 2 like 2,16E‐07 −1,410 215990_s_at BCL6 BCL6 transcription repressor 1,74E‐07 −1,700 200776_s_at BZW1 basic leucine zipper and W2 domains 1 1,09E‐06 −1,570 222309_at