Evidence for Positive Selection on Recent Human Transposable Element T Insertions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Proteome Profiling of Developing Murine Lens Through Mass
Lens Proteome Profiling of Developing Murine Lens Through Mass Spectrometry Shahid Y. Khan,1 Muhammad Ali,1 Firoz Kabir,1 Santosh Renuse,2 Chan Hyun Na,2 C. Conover Talbot Jr,3 Sean F. Hackett,1 and S. Amer Riazuddin1 1The Wilmer Eye Institute, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States 2Department of Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States 3Institute for Basic Biomedical Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States Correspondence: S. Amer Riazuddin, PURPOSE. We previously completed a comprehensive profile of the mouse lens transcriptome. The Wilmer Eye Institute, Johns Here, we investigate the proteome of the mouse lens through mass spectrometry–based Hopkins University School of Medi- protein sequencing at the same embryonic and postnatal time points. cine, 600 N. Wolfe Street; Maumenee 840, Baltimore, MD 21287, USA; METHODS. We extracted mouse lenses at embryonic day 15 (E15) and 18 (E18) and postnatal [email protected]. day 0 (P0), 3 (P3), 6 (P6), and 9 (P9). The lenses from each time point were preserved in three Submitted: February 2, 2017 distinct pools to serve as biological replicates for each developmental stage. The total cellular Accepted: October 13, 2017 protein was extracted from the lens, digested with trypsin, and labeled with isobaric tandem mass tags (TMT) for three independent TMT experiments. Citation: Khan SY, Ali M, Kabir F, et al. Proteome profiling of developing mu- RESULTS. A total of 5404 proteins were identified in the mouse ocular lens in at least one rine lens through mass spectrometry. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Region Based Gene Expression Via Reanalysis of Publicly Available Microarray Data Sets
University of Louisville ThinkIR: The University of Louisville's Institutional Repository Electronic Theses and Dissertations 5-2018 Region based gene expression via reanalysis of publicly available microarray data sets. Ernur Saka University of Louisville Follow this and additional works at: https://ir.library.louisville.edu/etd Part of the Bioinformatics Commons, Computational Biology Commons, and the Other Computer Sciences Commons Recommended Citation Saka, Ernur, "Region based gene expression via reanalysis of publicly available microarray data sets." (2018). Electronic Theses and Dissertations. Paper 2902. https://doi.org/10.18297/etd/2902 This Doctoral Dissertation is brought to you for free and open access by ThinkIR: The University of Louisville's Institutional Repository. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of ThinkIR: The University of Louisville's Institutional Repository. This title appears here courtesy of the author, who has retained all other copyrights. For more information, please contact [email protected]. REGION BASED GENE EXPRESSION VIA REANALYSIS OF PUBLICLY AVAILABLE MICROARRAY DATA SETS By Ernur Saka B.S. (CEng), University of Dokuz Eylul, Turkey, 2008 M.S., University of Louisville, USA, 2011 A Dissertation Submitted To the J. B. Speed School of Engineering in Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Computer Science and Engineering Department of Computer Engineering and Computer Science University of Louisville Louisville, Kentucky May 2018 Copyright 2018 by Ernur Saka All rights reserved REGION BASED GENE EXPRESSION VIA REANALYSIS OF PUBLICLY AVAILABLE MICROARRAY DATA SETS By Ernur Saka B.S. (CEng), University of Dokuz Eylul, Turkey, 2008 M.S., University of Louisville, USA, 2011 A Dissertation Approved On April 20, 2018 by the following Committee __________________________________ Dissertation Director Dr. -

CRYZ Conjugated Antibody
Product Datasheet CRYZ Conjugated Antibody Catalog No: #C27817 Package Size: #C27817-AF350 100ul #C27817-AF405 100ul #C27817-AF488 100ul Orders: [email protected] Support: [email protected] #C27817-AF555 100ul #C27817-AF594 100ul #C27817-AF647 100ul #C27817-AF680 100ul #C27817-AF750 100ul #C27817-Biotin 100ul Description Product Name CRYZ Conjugated Antibody Host Species Rabbit Clonality Polyclonal Isotype IgG Purification Affinity purification Applications most applications Species Reactivity Hu,Ms,Rt Immunogen Description Recombinant fusion protein of human CRYZ (NP_001123514.1). Conjugates Biotin AF350 AF405 AF488 AF555 AF594 AF647 AF680 AF750 Other Names CRYZ; crystallin zeta Accession No. Swiss-Prot#:Q08257NCBI Gene ID:1429 Calculated MW 37kDa Formulation 0.01M Sodium Phosphate, 0.25M NaCl, pH 7.6, 5mg/ml Bovine Serum Albumin, 0.02% Sodium Azide Storage Store at 4°C in dark for 6 months Application Details Suggested Dilution: AF350 conjugated: most applications: 1: 50 - 1: 250 AF405 conjugated: most applications: 1: 50 - 1: 250 AF488 conjugated: most applications: 1: 50 - 1: 250 AF555 conjugated: most applications: 1: 50 - 1: 250 AF594 conjugated: most applications: 1: 50 - 1: 250 AF647 conjugated: most applications: 1: 50 - 1: 250 AF680 conjugated: most applications: 1: 50 - 1: 250 AF750 conjugated: most applications: 1: 50 - 1: 250 Biotin conjugated: working with enzyme-conjugated streptavidin, most applications: 1: 50 - 1: 1,000 Background Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. The former class is also called phylogenetically-restricted crystallins. -

Rabbit Anti-CRYZ Antibody-SL14080R
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-CRYZ antibody SL14080R Product Name: CRYZ Chinese Name: 晶状体蛋白ζ抗体 Crystallin zeta (quinone reductase); Crystallin zeta; Cryz; DKFZp779E0834; Alias: FLJ41475; NADPH:quinone reductase; QOR_HUMAN; Quinone oxidoreductase; Zeta crystallin; Zeta-crystallin; Organism Species: Rabbit Clonality: Polyclonal React Species: Human, WB=1:500-2000ELISA=1:500-1000IHC-P=1:400-800IHC-F=1:400-800ICC=1:100- 500IF=1:100-500(Paraffin sections need antigen repair) Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 35kDa Cellular localization: cytoplasmic Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human CRYZ:61-160/329 Lsotype: IgGwww.sunlongbiotech.com Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. PubMed: PubMed Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. The latter class constitutes the major proteins of vertebrate eye lens and maintains the Product Detail: transparency and refractive index of the lens. The former class is also called phylogenetically-restricted crystallins. -

Mouse Cryz Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Cryz Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Cryz conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Cryz gene (NCBI Reference Sequence: NM_009968 ; Ensembl: ENSMUSG00000028199 ) is located on Mouse chromosome 3. 9 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 9 (Transcript: ENSMUST00000029850). Exon 5 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Cryz gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-5D19 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 5 starts from about 43.2% of the coding region. The knockout of Exon 5 will result in frameshift of the gene. The size of intron 4 for 5'-loxP site insertion: 2248 bp, and the size of intron 5 for 3'-loxP site insertion: 4586 bp. The size of effective cKO region: ~552 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 4 5 9 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Cryz Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Vast Human-Specific Delay in Cortical Ontogenesis Associated With
Supplementary information Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques Supplementary Methods Sample collection We used prefrontal cortex (PFC) and cerebellar cortex (CBC) samples from postmortem brains of 33 human (aged 0-98 years), 14 chimpanzee (aged 0-44 years) and 44 rhesus macaque individuals (aged 0-28 years) (Table S1). Human samples were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, USA, the Netherlands Brain Bank, Amsterdam, Netherlands and the Chinese Brain Bank Center, Wuhan, China. Informed consent for use of human tissues for research was obtained in writing from all donors or their next of kin. All subjects were defined as normal by forensic pathologists at the corresponding brain bank. All subjects suffered sudden death with no prolonged agonal state. Chimpanzee samples were obtained from the Yerkes Primate Center, GA, USA, the Anthropological Institute & Museum of the University of Zürich-Irchel, Switzerland and the Biomedical Primate Research Centre, Netherlands (eight Western chimpanzees, one Central/Eastern and five of unknown origin). Rhesus macaque samples were obtained from the Suzhou Experimental Animal Center, China. All non-human primates used in this study suffered sudden deaths for reasons other than their participation in this study and without any relation to the tissue used. CBC dissections were made from the cerebellar cortex. PFC dissections were made from the frontal part of the superior frontal gyrus. All samples contained an approximately 2:1 grey matter to white matter volume ratio. RNA microarray hybridization RNA isolation, hybridization to microarrays, and data preprocessing were performed as described previously (Khaitovich et al. -
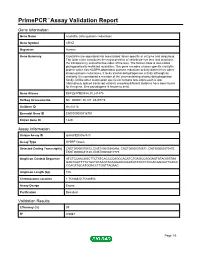
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name crystallin, zeta (quinone reductase) Gene Symbol CRYZ Organism Human Gene Summary Crystallins are separated into two classes: taxon-specific or enzyme and ubiquitous. The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. The former class is also called phylogenetically-restricted crystallins. This gene encodes a taxon-specific crystallin protein which has NADPH-dependent quinone reductase activity distinct from other known quinone reductases. It lacks alcohol dehydrogenase activity although by similarity it is considered a member of the zinc-containing alcohol dehydrogenase family. Unlike other mammalian species in humans lens expression is low. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. One pseudogene is known to exist. Gene Aliases DKFZp779E0834, FLJ41475 RefSeq Accession No. NC_000001.10, NT_032977.9 UniGene ID Hs.83114 Ensembl Gene ID ENSG00000116791 Entrez Gene ID 1429 Assay Information Unique Assay ID qHsaCED0057473 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000370872, ENST00000340866, ENST00000370871, ENST00000370870, ENST00000441120, ENST00000417775 Amplicon Context Sequence ATCTCCAACAGCTTCTATCACCCCAGCCACATCTGAGCCAGGAGTATAGGGTAA GAGTGGTTTTCTACTATAAGTACCAGAGCGAATGTATGTCTCCACGGGGTTGACA CCACATGCATGGACCTTGATTAGAAC Amplicon Length (bp) 105 Chromosome Location 1:75188820-75188954 Assay Design Exonic Purification Desalted -
![[KO Validated] CRYZ Rabbit Pab](https://docslib.b-cdn.net/cover/3896/ko-validated-cryz-rabbit-pab-5243896.webp)
[KO Validated] CRYZ Rabbit Pab
Leader in Biomolecular Solutions for Life Science [KO Validated] CRYZ Rabbit pAb Catalog No.: A19997 KO Validated Basic Information Background Catalog No. Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. A19997 The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. The former class is also called Observed MW phylogenetically-restricted crystallins. This gene encodes a taxon-specific crystallin 35KDa protein which has NADPH-dependent quinone reductase activity distinct from other known quinone reductases. It lacks alcohol dehydrogenase activity although by Calculated MW similarity it is considered a member of the zinc-containing alcohol dehydrogenase 20kDa/31kDa/35kDa family. Unlike other mammalian species, in humans, lens expression is low. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Category One pseudogene is known to exist. Primary antibody Applications WB Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 1429 Q08257 Immunogen Recombinant protein of human CRYZ. Synonyms CRYZ Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts from normal (control) and CRYZ knockout (KO) HeLa cells, using CRYZ antibody (A19997) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). -

Network Pharmacology-Based Approach Uncovers the Mechanism of Guanxinning Tablet for Treating Thrombus by Mapks Signal Pathway
ORIGINAL RESEARCH published: 13 May 2020 doi: 10.3389/fphar.2020.00652 Network Pharmacology-Based Approach Uncovers the Mechanism of GuanXinNing Tablet for Treating Thrombus by MAPKs Signal Pathway † † Mu-Lan Wang 1,2 , Qin-Qin Yang 1,3 , Xu-Hui Ying 2, Yuan-Yuan Li 1, Yang-Sheng Wu 1, Qi-Yang Shou 1, Quan-Xin Ma 1, Zi-Wei Zhu 2 and Min-Li Chen 1* 1 Academy of Chinese Medicine & Institute of Comparative Medicine, Zhejiang Chinese Medical University, Hangzhou, China, 2 The Department of Medicine, Chiatai Qingchunbao Pharmaceutical Co., Ltd., Hangzhou, China, 3 Department of Experimental Animals, Zhejiang Academy of Traditional Chinese Medicine, Hangzhou, China Edited by: Background: GuanXinNing tablet (GXNT), a traditional Chinese patent medicine, has Jianxun Liu, been found to have remarkable antithrombotic effects and can effectively inhibit pro- China Academy of Chinese Medical Sciences, China thrombotic factors in previous studies. However, the mechanism of its antithrombotic Reviewed by: effects remains little known. Songxiao Xu, fi fi Artron BioResearch Inc., Canada Methods: In this study, we rst determined and identi ed the sources of each main Yi Ding, compound in GXNT using liquid chromatography-mass spectrometry (LC-MS). Through Fourth Military Medical the approach of network pharmacology, we predicted the action targets of the active University, China Yunyao Jiang, components, mapped the target genes related to thrombus, and obtained potential Tsinghua University, China antithrombotic targets for active ingredients. We then performed gene ontology (GO) *Correspondence: enrichment analyses and KEGG signaling pathway analyses for the action targets, and Min-Li Chen – – – [email protected] constructed networks of active component target and active component target †These authors have contributed pathway for GXNT. -

Genome-Wide Association Analysis Identifies TYW3
Human Molecular Genetics, 2012, Vol. 21, No. 21 4774–4780 doi:10.1093/hmg/dds300 Advance Access published on July 26, 2012 Genome-wide association analysis identifies TYW3/CRYZ and NDST4 loci associated with circulating resistin levels Qibin Qi1,{, Claudia Menzaghi3,{, Shelly Smith4, Liming Liang2, Nathalie de Rekeneire7, Melissa E. Garcia6, Kurt K. Lohman4, Iva Miljkovic8, Elsa S. Strotmeyer8, Steve R. Cummings9, Alka M. Kanaya9, Frances A. Tylavsky10, Suzanne Satterfield10, Jingzhong Ding5, Eric B. Rimm1,2,11, Vincenzo Trischitta3,12, Frank B. Hu1,2,11, Yongmei Liu4,∗,{ and Lu Qi1,11,∗,{ 1Department of Nutrition and 2Department of Epidemiology, Harvard School of Public Health, Boston, MA, USA, 3Research Unit of Diabetes and Endocrine Diseases, IRCCS ‘Casa Sollievo della Sofferenza’, San Giovanni Rotondo, Italy, 4Division of Public Health Sciences and 5Department of Internal Medicine, Wake Forest School of Medicine, Downloaded from Winston-Salem, NC, USA, 6Laboratory of Epidemiology, Demography and Biometry, National Institute on Aging, Bethesda, MD, USA, 7Department of Geriatrics, Yale University School of Medicine, New Haven, CT, USA, 8Department of Epidemiology, University of Pittsburgh, Pittsburgh, PA, USA, 9Division of General Internal Medicine, University of California, San Francisco, CA, USA, 10Department of Preventive Medicine, University of Tennessee, Memphis, TN, USA, 11Channing Laboratory, Department of Medicine, Harvard Medical School, Boston, MA, USA http://hmg.oxfordjournals.org/ and 12Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy Received February 2, 2012; Revised July 10, 2012; Accepted July 19, 2012 Resistin is a polypeptide hormone that was reported to be associated with insulin resistance, inflammation and risk of type 2 diabetes and cardiovascular disease.