Redrawing the Ramachandran Plot After Inclusion of Hydrogen-Bonding
Total Page:16
File Type:pdf, Size:1020Kb
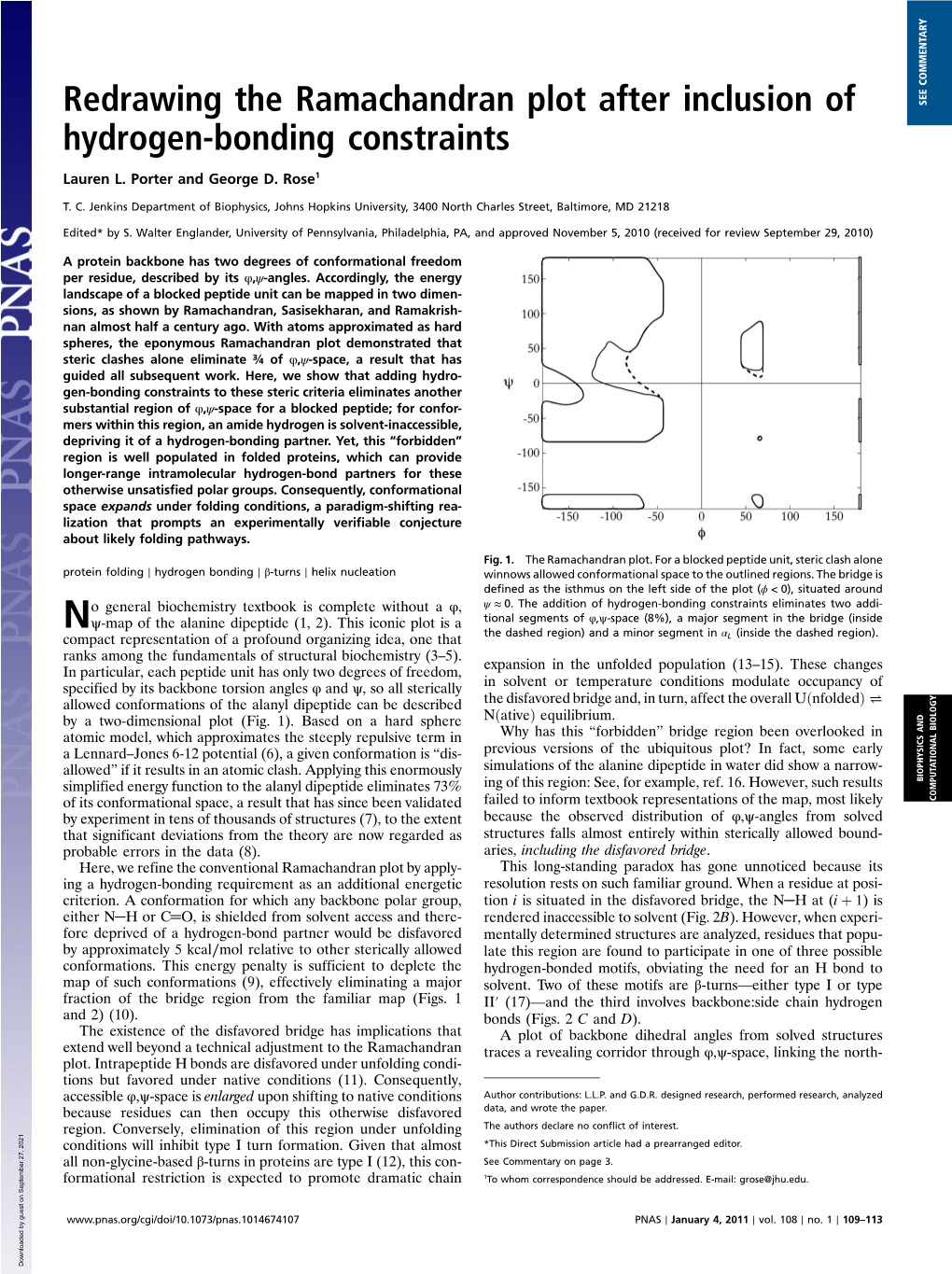
Load more
Recommended publications
-

Biomolecules
CHAPTER 3 Biomolecules 3.1 Carbohydrates In the previous chapter you have learnt about the cell and 3.2 Fatty Acids and its organelles. Each organelle has distinct structure and Lipids therefore performs different function. For example, cell membrane is made up of lipids and proteins. Cell wall is 3.3 Amino Acids made up of carbohydrates. Chromosomes are made up of 3.4 Protein Structure protein and nucleic acid, i.e., DNA and ribosomes are made 3.5 Nucleic Acids up of protein and nucleic acids, i.e., RNA. These ingredients of cellular organelles are also called macromolecules or biomolecules. There are four major types of biomolecules— carbohydrates, proteins, lipids and nucleic acids. Apart from being structural entities of the cell, these biomolecules play important functions in cellular processes. In this chapter you will study the structure and functions of these biomolecules. 3.1 CARBOHYDRATES Carbohydrates are one of the most abundant classes of biomolecules in nature and found widely distributed in all life forms. Chemically, they are aldehyde and ketone derivatives of the polyhydric alcohols. Major role of carbohydrates in living organisms is to function as a primary source of energy. These molecules also serve as energy stores, 2021-22 Chapter 3 Carbohydrade Final 30.018.2018.indd 50 11/14/2019 10:11:16 AM 51 BIOMOLECULES metabolic intermediates, and one of the major components of bacterial and plant cell wall. Also, these are part of DNA and RNA, which you will study later in this chapter. The cell walls of bacteria and plants are made up of polymers of carbohydrates. -

Density Estimation for Protein Conformation Angles Using a Bivariate Von Mises Distribution and Bayesian Nonparametrics
Density Estimation for Protein Conformation Angles Using a Bivariate von Mises Distribution and Bayesian Nonparametrics Kristin P. LENNOX, David B. DAHL, Marina VANNUCCI, and Jerry W. TSAI Interest in predicting protein backbone conformational angles has prompted the development of modeling and inference procedures for bivariate angular distributions. We present a Bayesian approach to density estimation for bivariate angular data that uses a Dirichlet process mixture model and a bivariate von Mises distribution. We derive the necessary full conditional distributions to fit the model, as well as the details for sampling from the posterior predictive distribution. We show how our density estimation method makes it possible to improve current approaches for protein structure prediction by comparing the performance of the so-called ‘‘whole’’ and ‘‘half’’ position dis- tributions. Current methods in the field are based on whole position distributions, as density estimation for the half positions requires techniques, such as ours, that can provide good estimates for small datasets. With our method we are able to demonstrate that half position data provides a better approximation for the distribution of conformational angles at a given sequence position, therefore providing increased efficiency and accuracy in structure prediction. KEY WORDS: Angular data; Density estimation; Dirichlet process mixture model; Torsion angles; von Mises distribution. 1. INTRODUCTION and Ohlson 2002; Lovell et al. 2003; Rother, Sapiro, and Pande 2008), but they behave poorly for these subdivided datasets. Computational structural genomics has emerged as a pow- This is unfortunate, because these subsets provide structure erful tool for better understanding protein structure and func- prediction that is more accurate, as it utilizes more specific tion using the wealth of data from ongoing genome projects. -

Structure Validation Article
14 STRUCTURAL QUALITY ASSURANCE Roman A. Laskowski The experimentally determined three-dimensional (3D) structures of proteins and nucleic acids represent the knowledge base from which so much understanding of biological processes has been derived over the last three decades of the twentieth cen- tury. Individual structures have provided explanations of specific biochemical functions and mechanisms, while comparisons of structures have given insights into general principles governing these complex molecules, the interactions they make, and their biological roles. The 3D structures form the foundation of structural bioinformatics; all structural analyses depend on them and would be impossible without them. Therefore, it is crucial to bear in mind two important truths about these structures, both of which result from the fact that they have been determined experimentally. The first is that the result of any experiment is merely a model that aims to give as good an explanation for the experimental data as possible. The term structure is commonly used, but you should realize that this should be correctly read as model. As such the model may be an accurate and meaningful representation of the molecule, or it may be a poor one. The quality of the data and the care with which the experiment has been performed will determine which it is. Independently performed experiments can arrive at very similar models of the same molecule; this suggests that both are accurate representations, that they are good models. The second important truth is that any experiment, however carefully performed, will have errors associated with it. These errors come in two distinct varieties: sys- tematic and random. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Prediction of Amino Acid Side Chain Conformation Using a Deep Neural
Prediction of amino acid side chain conformation using a deep neural network Ke Liu 1, Xiangyan Sun3, Jun Ma3, Zhenyu Zhou3, Qilin Dong4, Shengwen Peng3, Junqiu Wu3, Suocheng Tan3, Günter Blobel2, and Jie Fan1, Corresponding author details: Jie Fan, Accutar Biotechnology, 760 Parkside Ave, Room 213 Brooklyn, NY 11226, USA. [email protected] 1. Accutar Biotechnology, 760 parkside Ave, Room 213, Brooklyn, NY 11226, USA. 2. Laboratory of Cell Biology, Howard Hughes Medical Institute, The Rockefeller University, New York, New York 10065, USA 3. Accutar Biotechnology (Shanghai) Room 307, No. 6 Building, 898 Halei Rd, Shanghai, China 4. Fudan University 220 Handan Rd, Shanghai, China Summary: A deep neural network based architecture was constructed to predict amino acid side chain conformation with unprecedented accuracy. Amino acid side chain conformation prediction is essential for protein homology modeling and protein design. Current widely-adopted methods use physics-based energy functions to evaluate side chain conformation. Here, using a deep neural network architecture without physics-based assumptions, we have demonstrated that side chain conformation prediction accuracy can be improved by more than 25%, especially for aromatic residues compared with current standard methods. More strikingly, the prediction method presented here is robust enough to identify individual conformational outliers from high resolution structures in a protein data bank without providing its structural factors. We envisage that our amino acid side chain predictor could be used as a quality check step for future protein structure model validation and many other potential applications such as side chain assignment in Cryo-electron microscopy, crystallography model auto-building, protein folding and small molecule ligand docking. -

NIH Public Access Author Manuscript Proteins
NIH Public Access Author Manuscript Proteins. Author manuscript; available in PMC 2009 May 11. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Proteins. 2008 June ; 71(4): 1637±1646. doi:10.1002/prot.21845. Using Quantum Mechanics to Improve Estimates of Amino Acid Side Chain Rotamer Energies P. Douglas Renfrew, Glenn Butterfoss, and Brian Kuhlman Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27516, Email: [email protected] Abstract Amino acid side chains adopt a discrete set of favorable conformations typically referred to as rotamers. The relative energies of rotamers partially determine which side chain conformations are more often observed in protein structures and accurate estimates of these energies are important for predicting protein structure and designing new proteins. Protein modelers typically calculate side chain rotamer energies by using molecular mechanics (MM) potentials or by converting rotamer probabilities from the protein database (PDB) into relative free energies. One limitation of the knowledge-based energies is that rotamer preferences observed in the PDB can reflect internal side chain energies as well as longer-range interactions with the rest of the protein. Here, we test an alternative approach for calculating rotamer energies. We use three different quantum mechanics (QM) methods (second order Moller-Plesset (MP2), density functional theory (DFT) energy calculation using the B3LYP functional, and Hartree-Fock) to calculate the energy of amino acid rotamers in a dipeptide model system, and then use these pre-calculated values in side chain placement simulations. Energies were calculated for over 35,000 different conformations of leucine, isoleucine and valine dipeptides with backbone torsion angles from the helical and strand regions of the Ramachandran plot. -

Theoretical Protein Structure Prediction of Glucagon-Like Peptide 2 Receptor Using Homology Modelling
J. Chosun Natural Sci. Vol. 10, No. 3 (2017) pp. 119 − 124 https://doi.org/10.13160/ricns.2017.10.3.119 Theoretical Protein Structure Prediction of Glucagon-like Peptide 2 Receptor Using Homology Modelling Santhosh Kumar Nagarajan1,2 and Thirumurthy Madhavan2† Abstract Glucagon-like peptide 2 receptor, a GPCR, binds with the glucagon-like peptide, GLP-2 and regulates various metabolic functions in the gastrointestinal tract. It plays an important role in the nutrient homeostasis related to nutrient assimilation by regulating mucosal epithelium. GLP-2 receptor affects the cellular response to external injury, by controlling the intestinal crypt cell proliferation. As they are therapeutically attractive towards diseases related with the gastrointestinal tract, it becomes essential to analyse their structural features to study the pathophysiology of the diseases. As the three dimensional structure of the protein is not available, in this study, we have performed the homology modelling of the receptor based on single- and multiple template modeling. The models were subjected to model validation and a reliable model based on the validation statistics was identified. The predicted model could be useful in studying the structural features of GLP-2 receptor and their role in various diseases related to them. Keywords: GLP2 Receptor, GPCR, GLP2, Homology Modelling. 1. Introduction nutrient homeostasis proximal to nutrient assimilation by controlling the stasis of mucosal epithelium. The sig- Glucagon and glucagon like peptides (GLPs) are nalling via GLP-2 receptor regulates the intestinal crypt secreted in the pancreas, gut, CNS and PNS and control cell proliferation, directly affecting the cellular response various metabolic functions[1]. -

Protein Structure Prediction and Potential Energy Landscape Analysis Using Continuous Global Minimization
Protein Structure Prediction and Potential Energy Landscape Analysis using Continuous Global Minimization KEN A. DILL Departmentof Pharmaceutical Chemistry, University of California at San Francisco, San Francisco, CA 94118 <[email protected]~edu~ ANDREW T. PHILLIPS ComputerScience Department, United StatesNaval Academy,Annapolis, MD 21402 <[email protected]> J. BEN ROSEN Computer Science and Engineering Department, University of California at San Diego, San Diego, CA 92093 <[email protected]> INTRODUCTION form of energy minimization technique, such as simu- lated annealing and genetic algorithms, rather than the Proteins require specific three-dimensional conforma- computationally more efficient continuous minimiza- tions to function properly. These “native” conforma- tion techniques. Each such method correctly predicts a tions result primarily from intramolecular interactions few protein structures, but missesmany others, and the between the atoms in the macromolecule, and also process is both slow and limited to structures of rela- intermolecular interactions between the macromole- tively small size. cule and the surrounding solvent. Although the folding The purpose of this paper is to show how one can process can be quite complex, the instructions guiding apply more efficient continuous minimization tech- this process are specified by the one-dimensional pri- niques to the energy minimization problem by using mary sequenceof the protein or nucleic acid: external an accurate continuous approximation to the discrete factors, such as helper (chaperone) proteins, present at information provided for known protein structures. In the time of folding have no effect on the final state of addition, we will show how the results of one particu- the protein. Many denatured proteins spontaneously lar computational method for protein structure predic- refold into functional conformations once denaturing tion (the CGU algorithm), which is based on this conditions are removed. -

Three Dimensional Structure Modeling and Ramachandran Plot Analysis of Autographa Californica Nucleopolyhdro Viral Protein
Gopukumar et al (2020): Analysis of nucleopolyhedro viral protein March 2020 Vol. 23 Issue S6 Three dimensional structure modeling and ramachandran plot analysis of autographa californica nucleopolyhdro viral protein S.T. Gopukumar1*, Sreeya G. Nair2, R. Radha2, N.V. Sugathan3, Anooj E. S4, Lekshmi Gangadhar4 1Department of Medical Research, Sarada Krishna Homoeopathic Medical College, Kulasekharam. TN, India 2Department of Zoology, Sree Ayyappa College for Women, Chunkankadai, Nagercoil, TN, India 3Department of Practice of Medicine, Sarada Krishna Homoeopathic Medical College, Kulasekharam. TN, India 4Xcellogen Biotech India Pvt Ltd, Nagercoil, Tamilnadu, India *Corresponding Author E-mail: [email protected] (Gopukuma) ABSTRACT: Background: Autographa Californica’s nucleopolyhedro is a viral protein found in baciloviridae. It is made of double stranded DNA genome. They viral protein easily infects this insect and affects the expression system. Method: The main aim of this work is to predict various viral proteins (Autographa Californica nucleopolyhedro). FASTA sequences are retrived from NCBI (National Center for Biotechnology Information) database. Then Basic Local Alignment Search Tool (BLAST) used to find regions of similarity in different sequences. The homology modeling was done in swiss model. The template model also constructed. Results: Comparative modeling of this program to predict the protein sequence identity is 46.83%.The template protein e value is 110 and they chain A having identify 100% similarity maximum score 320.Further the protein structure was validated in Ramachandran plot. Conclusion: From Ramachandran plot analysis it was found that the portions of residues falling into the favored regions was 98%.The predicted 3D model further characterized and analyzed using other techniques. -

Molecular Modeling 2021
Molecular Modeling 2021 A course about proteins protein conformation protein shape Protein energy protein interactions protein dynamics protein drug interactions Administrative • Course website: on Blackboard learn: https://lms.rpi.edu/ • Office hour: 4 – 5 PM Wednesdays, on webex • Grading policy – Assignment and quiz (30 % total) – midterm (20%) – final (20%) – Presentation (20%) – Class participation (answering questions in class) and attendance (10%) • Questions ALWAYS welcome! Syllabus Installing MOE • Download MOE to your personal computer using file on RPI box https://rpi.box.com/s/ mdjcw69wffo5roe5p7nse4n16wznf97u !! If your RPI box account is not set up, use this link to set it up: https://itssc.rpi.edu/hc/en-us/articles/ 360004829931-Requesting-a-Box-RPI-Account • Copy the license file (license.dat in the box directory) into the installed MOE local directory • To run MOE, you need to access the license server located on RPI campus: got to have VPN running! – Install “cisco anyconnec”t: https://itssc.rpi.edu/hc/en-us/ articles/360008783172-VPN-Installation-and-Connection Instructor • Chunyu Wang • Associate Professor of Biological Sciences • Expert in NMR, protein structure, Alzheimer's disease, Inteins and Hedgehog autoprocessing • Hobbies: photography, astronomy, astrophotography, tennis, bridge, gardening, swimming etc. NMR structure of Pab PolII intein: Hint fold Cover of Nov.4th 2011 Issue of JBC Hint = Hedgehog Intein domain/fold Du et.al. J. Biol. Chem., 2011, 286, 38638-38648. 6 What are Aβ, Αβ40 and Αβ42 ? Aβ42 Aβ40 Major component of the amyloid plaque Aβ40 and Aβ42 are major species of Aβ Aβ42 aggregates faster and is more toxic than Aβ40 In familial Alzheimer’s disease (FAD), Aβ42/Aβ40 ↑ Aβ40 Aβ42 . -

Welcome to Week 2 Chapter 3 – Protein Structure 3.1 Intro To
Welcome to Week 2 Starting week two video Please watch the online video (1 minutes 24 seconds). OPTIONAL‐Please participate in the online discussion forum. Chapter 3 – Protein Structure Introduction to Chapter 3 Chapter 3 contains two subsections. Intro to Structure Part 1 Amino Acids, Primary Structure, and Secondary Structure Intro to Structure Part 2 Tertiary Structure, Quaternary Structure, and X‐Ray Crystallography At the conclusion of this chapter, you should understand how the ordering of individual amino acids in a protein can affect the localized and global folding and function of the entire protein. You should further have some appreciation of how x‐ray crystallographic data is used to determine the structure of proteins. OPTIONAL‐Please participate in the online discussion forum. 3.1 Intro to Structure Pt 1 Amino acids to secondary structure video Please watch the online video (8 minutes, 15 seconds). A condensed summary of this video can be found in the Video summary page. OPTIONAL‐Please participate in the online discussion forum. Working with Protein Data Bank entries Background: The structural information freely available in the Protein Data Bank is immense. It allows anyone to be able to study protein structure and function. Instructions: Use the tools in the PDB to examine the structures mentioned below and answer the assessment questions. Learning Goals: To learn how to manipulate proteins and identify their structural elements with the tools in the PDB. Please complete the online exercise. OPTIONAL‐Please participate in the online discussion forum. Finding PDB entry codes Background: Searching a specific entry in the Protein Data Bank requires one to know the corresponding PDB code. -

Bioinformatics for Molecular Biology Structural Bioinformatics Tools, Predictors, and 3D Modeling – Structural Biology Review
Bioinformatics for molecular biology Structural bioinformatics tools, predictors, and 3D modeling – Structural Biology Review Dr Jon K. Lærdahl, Research Scientist Department of Microbiology, Oslo University Hospital - Rikshospitalet & Bioinformatics Core Facility/CLS initiative, University of Oslo E-mail: [email protected] Phone: +47 22844784 Group: Torbjørn Rognes (http://www.ous-research.no/rognes) CF: Bioinformatics services (http://core.rr-research.no/bioinformatics) CLS: Bioinformatics education (http://www.mn.uio.no/ifi/english/research/networks/clsi) Main research area: Structural and Applied Bioinformatics Jon K. Lærdahl, Structural Bioinformatics Now: Overview • Protein Structure Review – Amino acids, polypeptides, secondary structure elements, visualization, structure determination by X-ray crystallography and NMR methods, PDB Later… • Structure comparison and classification (CASP & SCOP) • Predictors • 3D structure modeling – Ab initio – Threading/fold recognition – Homology modeling • Practical exercises – PyMOL & visualization • Practical Exercises – Homology modeling of influenza neuraminidase (Tamiflu resistance?) – Other homology modeling – Threading – Your own project? Stop me and ask questions!! Jon K. Lærdahl, Amino acids – the building blocks Structural Bioinformatics Proteins are built from 20 naturally occurring amino of proteins acids. They have an amino (-NH2) and acidic The side chain group (R) determines the properties of the (-COOH) functional group amino acid Side chain Carboxyl group Amino Zwitterionic form