Introduction to Linux & UPPMAX
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UKUI: a Lightweight Desktop Environment Based on Pluggable
2016 International Conference on Artificial Intelligence and Computer Science (AICS 2016) ISBN: 978-1-60595-411-0 UKUI: A Lightweight Desktop Environment Based on Pluggable Framework for Linux Distribution Jie YU1, Lu SI1,*, Jun MA1, Lei LUO1, Xiao-dong LIU1, Ya-ting KUANG2, Huan PENG2, Rui LI1, Jin-zhu KONG2 and Qing-bo WU1 1College of Computer, National University of Defense Technology, Changsha, China 2Tianjin KYLIN Information Technology Co., Ltd, Tianjin, China *[email protected] *Corresponding author Keywords: Desktop environment, Ubuntu, User interface. Abstract. Ubuntu is an operating system with Linux kernel based on Debian and distributed as free and open-source software. It uses Unity as its default desktop environment, which results in more difficulties of usage for Microsoft Windows users. In this paper, we present a lightweight desktop environment named UKUI based on UbuntuKylin, the official Chinese version of Ubuntu, for Linux distribution. It is designed as a pluggable framework and provides better user experience during human-computer interaction. In order to evaluate the performance of UKUI, a set of testing bench suits were performed on a personal computer. Overall, the results showed that UKUI has better performance compared with Unity. Introduction Linux is a freely available operating system (OS) originated by Linux Torvalds and further developed by thousands of others. Typically, Linux is packaged in a form known as a Linux distribution for both desktop and server use. Some of the most popular mainstream Linux distributions are Red Hat [1], Ubuntu [2], Arch [3], openSUSY [4], Gentoo [5], etc. There are several desktop environments available for nowadays modern Linux distributions, such as XFCE [6], GNOME [7], KDE [8] and LXDE [9]. -

GNU Grep: Print Lines That Match Patterns Version 3.7, 8 August 2021
GNU Grep: Print lines that match patterns version 3.7, 8 August 2021 Alain Magloire et al. This manual is for grep, a pattern matching engine. Copyright c 1999{2002, 2005, 2008{2021 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.3 or any later version published by the Free Software Foundation; with no Invariant Sections, with no Front-Cover Texts, and with no Back-Cover Texts. A copy of the license is included in the section entitled \GNU Free Documentation License". i Table of Contents 1 Introduction ::::::::::::::::::::::::::::::::::::: 1 2 Invoking grep :::::::::::::::::::::::::::::::::::: 2 2.1 Command-line Options ::::::::::::::::::::::::::::::::::::::::: 2 2.1.1 Generic Program Information :::::::::::::::::::::::::::::: 2 2.1.2 Matching Control :::::::::::::::::::::::::::::::::::::::::: 2 2.1.3 General Output Control ::::::::::::::::::::::::::::::::::: 3 2.1.4 Output Line Prefix Control :::::::::::::::::::::::::::::::: 5 2.1.5 Context Line Control :::::::::::::::::::::::::::::::::::::: 6 2.1.6 File and Directory Selection:::::::::::::::::::::::::::::::: 7 2.1.7 Other Options ::::::::::::::::::::::::::::::::::::::::::::: 9 2.2 Environment Variables:::::::::::::::::::::::::::::::::::::::::: 9 2.3 Exit Status :::::::::::::::::::::::::::::::::::::::::::::::::::: 12 2.4 grep Programs :::::::::::::::::::::::::::::::::::::::::::::::: 13 3 Regular Expressions ::::::::::::::::::::::::::: 14 3.1 Fundamental Structure :::::::::::::::::::::::::::::::::::::::: -

Linux Tutorial Last Updated: September 29 2021 for Windows Users
VLAAMS SUPERCOMPUTER Innovative Computing CENTRUM for A Smarter Flanders Linux Tutorial Last updated: September 29 2021 For Windows Users Authors: Jasper Devreker (UGent), Ewan Higgs (UGent), Kenneth Hoste (UGent) Acknowledgement: VSCentrum.be Audience: This document is a hands-on guide for using the Linux command line in the context of the UGent HPC infrastructure. The command line (sometimes called ’shell’) can seems daunting at first, but with a little understanding can be very easy to use. Everything you do startsatthe prompt. Here you have the liberty to type in any commands you want. Soon, you will be able to move past the limited point and click interface and express interesting ideas to the computer using the shell. Gaining an understanding of the fundamentals of Linux will help accelerate your research using the HPC infrastructure. You will learn about commands, managing files, and some scripting basics. Notification: In$ commands this tutorial specific commands are separated from the accompanying text: These should be entered by the reader at a command line in a terminal on the UGent-HPC. They appear in all exercises preceded by a $ and printed in bold. You’ll find those actions ina grey frame. Button are menus, buttons or drop down boxes to be pressed or selected. “Directory” is the notation for directories (called “folders” in Windows terminology) or specific files. (e.g., “/user/home/gent/vsc400/vsc40000”) “Text” Is the notation for text to be entered. Tip: A “Tip” paragraph is used for remarks or tips. They can also be downloaded from the VSC website at https://www.vscentrum.be. -

Cygwin User's Guide
Cygwin User’s Guide Cygwin User’s Guide ii Copyright © Cygwin authors Permission is granted to make and distribute verbatim copies of this documentation provided the copyright notice and this per- mission notice are preserved on all copies. Permission is granted to copy and distribute modified versions of this documentation under the conditions for verbatim copying, provided that the entire resulting derived work is distributed under the terms of a permission notice identical to this one. Permission is granted to copy and distribute translations of this documentation into another language, under the above conditions for modified versions, except that this permission notice may be stated in a translation approved by the Free Software Foundation. Cygwin User’s Guide iii Contents 1 Cygwin Overview 1 1.1 What is it? . .1 1.2 Quick Start Guide for those more experienced with Windows . .1 1.3 Quick Start Guide for those more experienced with UNIX . .1 1.4 Are the Cygwin tools free software? . .2 1.5 A brief history of the Cygwin project . .2 1.6 Highlights of Cygwin Functionality . .3 1.6.1 Introduction . .3 1.6.2 Permissions and Security . .3 1.6.3 File Access . .3 1.6.4 Text Mode vs. Binary Mode . .4 1.6.5 ANSI C Library . .4 1.6.6 Process Creation . .5 1.6.6.1 Problems with process creation . .5 1.6.7 Signals . .6 1.6.8 Sockets . .6 1.6.9 Select . .7 1.7 What’s new and what changed in Cygwin . .7 1.7.1 What’s new and what changed in 3.2 . -

Scons API Docs Version 4.2
SCons API Docs version 4.2 SCons Project July 31, 2021 Contents SCons Project API Documentation 1 SCons package 1 Module contents 1 Subpackages 1 SCons.Node package 1 Submodules 1 SCons.Node.Alias module 1 SCons.Node.FS module 9 SCons.Node.Python module 68 Module contents 76 SCons.Platform package 85 Submodules 85 SCons.Platform.aix module 85 SCons.Platform.cygwin module 85 SCons.Platform.darwin module 86 SCons.Platform.hpux module 86 SCons.Platform.irix module 86 SCons.Platform.mingw module 86 SCons.Platform.os2 module 86 SCons.Platform.posix module 86 SCons.Platform.sunos module 86 SCons.Platform.virtualenv module 87 SCons.Platform.win32 module 87 Module contents 87 SCons.Scanner package 89 Submodules 89 SCons.Scanner.C module 89 SCons.Scanner.D module 93 SCons.Scanner.Dir module 93 SCons.Scanner.Fortran module 94 SCons.Scanner.IDL module 94 SCons.Scanner.LaTeX module 94 SCons.Scanner.Prog module 96 SCons.Scanner.RC module 96 SCons.Scanner.SWIG module 96 Module contents 96 SCons.Script package 99 Submodules 99 SCons.Script.Interactive module 99 SCons.Script.Main module 101 SCons.Script.SConsOptions module 108 SCons.Script.SConscript module 115 Module contents 122 SCons.Tool package 123 Module contents 123 SCons.Variables package 125 Submodules 125 SCons.Variables.BoolVariable module 125 SCons.Variables.EnumVariable module 125 SCons.Variables.ListVariable module 126 SCons.Variables.PackageVariable module 126 SCons.Variables.PathVariable module 127 Module contents 127 SCons.compat package 129 Module contents 129 Submodules 129 SCons.Action -

Aspera CLI User Guide
Aspera Command- Line Interface Guide 3.7.7 Mac OS X Revision: 74 Generated: 09/25/2018 16:52 Contents Introduction............................................................................................................... 3 System Requirements............................................................................................... 3 Installation................................................................................................................. 3 Installing the Aspera CLI.....................................................................................................................................3 Configuring for Faspex.........................................................................................................................................4 Configuring for Aspera on Cloud........................................................................................................................ 4 Uninstalling........................................................................................................................................................... 5 aspera: The Command-Line Transfer Client........................................................ 5 About the Command-Line Client.........................................................................................................................5 Prerequisites.......................................................................................................................................................... 6 aspera Command Reference................................................................................................................................ -

Bash Guide for Beginners
Bash Guide for Beginners Machtelt Garrels Garrels BVBA <tille wants no spam _at_ garrels dot be> Version 1.11 Last updated 20081227 Edition Bash Guide for Beginners Table of Contents Introduction.........................................................................................................................................................1 1. Why this guide?...................................................................................................................................1 2. Who should read this book?.................................................................................................................1 3. New versions, translations and availability.........................................................................................2 4. Revision History..................................................................................................................................2 5. Contributions.......................................................................................................................................3 6. Feedback..............................................................................................................................................3 7. Copyright information.........................................................................................................................3 8. What do you need?...............................................................................................................................4 9. Conventions used in this -

Lab Work 06. Linux Shell. Files Globbing & Streams Redirection
LAB WORK 06. LINUX SHELL. FILES GLOBBING & STREAMS REDIRECTION. 1. PURPOSE OF WORK • Learn to use shell file globbing (wildcard); • Learn basic concepts about standard UNIX/Linux streams redirections; • Acquire skills of working with filter-programs. • Get experience in creating composite commands that have a different functional purpose than the original commands. 2. TASKS FOR WORK NOTE. Start Your UbuntuMini Virtual Machine on your VirtualBox. You need only Linux Terminal to complete the lab tasks. Before completing the tasks, make a Snapshot of your Virtual Linux. If there are problems, you can easily go back to working condition! 2.0. Create new User account for this Lab Work. • Login as student account (user with sudo permissions). • Create new user account, example stud. Use adduser command. (NOTE. You can use the command “userdel –rf stud” to delete stud account from your Linux.) $ sudo adduser stud • Logout from student account (logout) and login as stud. 2.1. Shell File Globbing Study. 2.2. File Globbing Practice. (Fill in a Table 1 and Table 2) 2.3. Command I/O Redirection Study. 2.4. Redirection Practice. (Fill in a Table 3 and Table 4) © Yuriy Shamshin, 2021 1/20 3. REPORT Make a report about this work and send it to the teacher’s email (use a docx Report Blank). REPORT FOR LAB WORK 06: LINUX SHELL. FILES GLOBBING & STREAMS REDIRECTION Student Name Surname Student ID (nV) Date 3.1. Insert Completing Table 1. File globbing understanding. 3.2. Insert Completing Table 2. File globbing creation. 3.3. Insert Completing Table 3. Command I/O redirection understanding. -
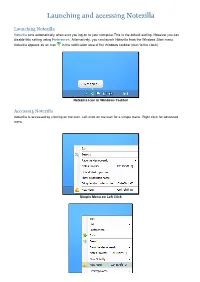
Launching and Accessing Notezilla
Launching and accessing Notezilla Launching Notezilla Notezilla runs automatically when ever you log on to your computer.This is the default setting. However you can disable this setting using Preferences. Alternatively, you can launch Notezilla from the Windows Start menu. Notezilla appears as an icon in the notification area of the Windows taskbar (next to the clock). Notezilla Icon in Windows Taskbar Accessing Notezilla Notezilla is accessed by clicking on the icon. Left click on the icon for a simple menu. Right click for advanced menu. Simple Menu on Left Click Advanced Menu on Right Click Writing your first sticky note Creating a sticky note Left click on the Notezilla icon in the Windows taskbar (next to the clock) and choose New Note from the menu to create a new sticky note. A sticky note appears on your Windows Desktop (see picture below). Right clicking on the Note Toolbar pops up the Note Menu . Sticky Note Type your tasks, appointments or a memo inside this sticky note. Setting a reminder To set a reminder, choose Due Date from the Note Menu. For faster access, use the shortcut key Ctrl+R. Positioning the sticky note You can change the position of the sticky note by simply holding the left mouse button on the note toolbar (or the note title) and dragging it to a different location on the desktop. Accessing the sticky note The sticky note that you created on the desktop may be overlapped by other program windows. You may need to access it again for viewing or editing. To view the note, left click on Notezilla icon and choose Bring Notes On Top. -

Lecture 10: File Systems File Systems, Databases, Cloud Storage
Lecture 10: File systems File systems, databases, cloud storage • file: a sequence of bytes stored on a computer – content is arbitrary (just bytes); any structure is imposed by the creator of the file, not by the operating system • file system: software that provides hierarchical storage and organization of files, usually on a single computer (or nearby) – a significant part of the operating system • database: an integrated collection of logically related records – data is organized and structured for efficient systematic access – may be distributed across lots of machines & geographically dispersed • database system: software that provides efficient access to information in a database – not usually part of the operating system • cloud storage: the same thing, but on someone else's computer(s) File systems: managing stored information • logical structure: users and programs see a hierarchy of folders (or directories) and files – a folder contains references to folder and files – "root" folder ultimately leads to all others – a file is just a sequence of bytes contents determined and interpreted by programs, not the operating system – a folder is a special file that contains names of other folders & files plus other information like size, time of change, etc. contents are completely controlled by the operating system • physical structure: disk drives operate in tracks, sectors, etc. – other storage devices have other physical properties • the operating system converts between these two views – does whatever is necessary to maintain the file/folder -

Bash Tutorial
Bash Shell Lecturer: Prof. Andrzej (AJ) Bieszczad Email: [email protected] Phone: 818-677-4954 Bash Shell The shell of Linux • Linux has a variety of different shells: – Bourne shell (sh), C shell (csh), Korn shell (ksh), TC shell (tcsh), Bour ne Again shell (bash). • Certainly the most popular shell is “bash”. Bash is an sh- compatible shell that incorporates useful features from the Korn shell (ksh) and C shell (csh). • It is intended to conform to the IEEE POSIX P1003.2/ISO 9945.2 Shell and Tools standard. • It offers functional improvements over sh for both programming and interactive use. Bash Shell Programming or Scripting ? • bash is not only an excellent command line shell, but a scripting language in itself. Shell scripting allows us to use the shell's abilities and to automate a lot of tasks that would otherwise require a lot of commands. • Difference between programming and scripting languages: – Programming languages are generally a lot more powerful and a lot faster than scriptin g languages. Programming languages generally start from source code and are compil ed into an executable. This executable is not easily ported into different operating syste ms. – A scripting language also starts from source code, but is not compiled into an executabl e. Rather, an interpreter reads the instructions in the source file and executes each inst ruction. Interpreted programs are generally slower than compiled programs. The main a dvantage is that you can easily port the source file to any operating system. bash is a s cripting language. Other examples of scripting languages are Perl, Lisp, and Tcl. -

Cygwin User's Guide
Cygwin User’s Guide i Cygwin User’s Guide Cygwin User’s Guide ii Copyright © 1998, 1999, 2000, 2001, 2002, 2003, 2004, 2005, 2006, 2007, 2008, 2009, 2010, 2011, 2012 Red Hat, Inc. Permission is granted to make and distribute verbatim copies of this documentation provided the copyright notice and this per- mission notice are preserved on all copies. Permission is granted to copy and distribute modified versions of this documentation under the conditions for verbatim copying, provided that the entire resulting derived work is distributed under the terms of a permission notice identical to this one. Permission is granted to copy and distribute translations of this documentation into another language, under the above conditions for modified versions, except that this permission notice may be stated in a translation approved by the Free Software Foundation. Cygwin User’s Guide iii Contents 1 Cygwin Overview 1 1.1 What is it? . .1 1.2 Quick Start Guide for those more experienced with Windows . .1 1.3 Quick Start Guide for those more experienced with UNIX . .1 1.4 Are the Cygwin tools free software? . .2 1.5 A brief history of the Cygwin project . .2 1.6 Highlights of Cygwin Functionality . .3 1.6.1 Introduction . .3 1.6.2 Permissions and Security . .3 1.6.3 File Access . .3 1.6.4 Text Mode vs. Binary Mode . .4 1.6.5 ANSI C Library . .5 1.6.6 Process Creation . .5 1.6.6.1 Problems with process creation . .5 1.6.7 Signals . .6 1.6.8 Sockets . .6 1.6.9 Select .