Mineralogy of Sulfides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Krásno Sn-W Ore District Near Horní Slavkov: Mining History, Geological and Mineralogical Characteristics
Journal of the Czech Geological Society 51/12(2006) 3 The Krásno Sn-W ore district near Horní Slavkov: mining history, geological and mineralogical characteristics Sn-W rudní revír Krásno u Horního Slavkova historie tìby, geologická a mineralogická charakteristika (47 figs, 1 tab) PAVEL BERAN1 JIØÍ SEJKORA2 1 Regional Museum Sokolov, Zámecká 1, Sokolov, CZ-356 00, Czech Republic 2 Department of Mineralogy and Petrology, National Museum, Václavské nám. 68, Prague 1, CZ-115 79, Czech Republic The tin-tungsten Krásno ore district near Horní Slavkov (Slavkovský les area, western Bohemia) belongs to the most important areas of ancient mining in the Czech Republic. The exceptionally rich and variable mineral associations, and the high number of mineral species, make this area one of the most remarkable mineralogical localities on a worldwide scale. The present paper reviews the data on geological setting of the ore district, individual ore deposits and mining history. Horní Slavkov and Krásno were known as a rich source of exquisite quality mineral specimens stored in numerous museum collections throughout Europe. The old museum specimens are often known under the German locality names of Schlaggenwald (= Horní Slavkov) and Schönfeld (=Krásno). The megascopic properties and paragenetic position of selected mineral classics are reviewed which include arsenopyrite, fluorapatite, fluorite, hübnerite, chalcopyrite, carpholite, cassiterite, quartz, molybdenite, rhodochrosite, sphalerite, topaz and scheelite. Key words: Sn-W ores; tin-tungsten mineralization; mining history; ore geology; mineralogy; Slavkovský les; Krásno, Horní Slavkov ore district; Czech Republic. Introduction valleys dissected parts of the area. In the ore district area, the detailed surface morphology is modified by large de- In the mining history of Central Europe, Bohemia and pressions caused by the collapse of old underground Moravia are known as important source of gold, silver, workings and by extensive dumps. -

Biofilm Adhesion on the Sulfide Mineral Bornite & Implications for Astrobiology
University of Rhode Island DigitalCommons@URI Open Access Master's Theses 2019 BIOFILM ADHESION ON THE SULFIDE MINERAL BORNITE & IMPLICATIONS FOR ASTROBIOLOGY Margaret M. Wilson University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/theses Recommended Citation Wilson, Margaret M., "BIOFILM ADHESION ON THE SULFIDE MINERAL BORNITE & IMPLICATIONS FOR ASTROBIOLOGY" (2019). Open Access Master's Theses. Paper 1517. https://digitalcommons.uri.edu/theses/1517 This Thesis is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Master's Theses by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. BIOFILM ADHESION ON THE SULFIDE MINERAL BORNITE & IMPLICATIONS FOR ASTROBIOLOGY BY MARGARET M. WILSON A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN BIOLOGICAL & ENVIRONMENTAL SCIENCE UNIVERSITY OF RHODE ISLAND 2019 MASTER OF SCIENCE IN BIOLOGICAL & ENVIRONMENTAL SCIENCE THESIS OF MARGARET M. WILSON APPROVED: Thesis Committee: Major Professor Dawn Cardace José Amador Roxanne Beinart Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2019 ABSTRACT We present research observing and documenting the model organism, Pseudomonas fluorescens (P. fluorescens), building biofilm on a natural mineral substrate composed largely of bornite (Cu5FeS4), a copper-iron sulfide mineral, with closely intergrown regions of covellite (CuS) and chalcopyrite (CuFeS2). In examining biofilm establishment on sulfide minerals, we investigate a potential habitable niche for microorganisms in extraterrestrial sites. Geochemical microenvironments on Earth and in the lab can also serve as analogs for important extraterrestrial sites, such as sheltered, subsurface microenvironments on Mars. -

LOW TEMPERATURE HYDROTHERMAL COPPER, NICKEL, and COBALT ARSENIDE and SULFIDE ORE FORMATION Nicholas Allin
Montana Tech Library Digital Commons @ Montana Tech Graduate Theses & Non-Theses Student Scholarship Spring 2019 EXPERIMENTAL INVESTIGATION OF THE THERMOCHEMICAL REDUCTION OF ARSENITE AND SULFATE: LOW TEMPERATURE HYDROTHERMAL COPPER, NICKEL, AND COBALT ARSENIDE AND SULFIDE ORE FORMATION Nicholas Allin Follow this and additional works at: https://digitalcommons.mtech.edu/grad_rsch Part of the Geotechnical Engineering Commons EXPERIMENTAL INVESTIGATION OF THE THERMOCHEMICAL REDUCTION OF ARSENITE AND SULFATE: LOW TEMPERATURE HYDROTHERMAL COPPER, NICKEL, AND COBALT ARSENIDE AND SULFIDE ORE FORMATION by Nicholas C. Allin A thesis submitted in partial fulfillment of the requirements for the degree of Masters in Geoscience: Geology Option Montana Technological University 2019 ii Abstract Experiments were conducted to determine the relative rates of reduction of aqueous sulfate and aqueous arsenite (As(OH)3,aq) using foils of copper, nickel, or cobalt as the reductant, at temperatures of 150ºC to 300ºC. At the highest temperature of 300°C, very limited sulfate reduction was observed with cobalt foil, but sulfate was reduced to sulfide by copper foil (precipitation of Cu2S (chalcocite)) and partly reduced by nickel foil (precipitation of NiS2 (vaesite) + NiSO4·xH2O). In the 300ºC arsenite reduction experiments, Cu3As (domeykite), Ni5As2, or CoAs (langisite) formed. In experiments where both sulfate and arsenite were present, some produced minerals were sulfarsenides, which contained both sulfide and arsenide, i.e. cobaltite (CoAsS). These experiments also produced large (~10 µm along longest axis) euhedral crystals of metal-sulfide that were either imbedded or grown upon a matrix of fine-grained metal-arsenides, or, in the case of cobalt, metal-sulfarsenide. Some experimental results did not show clear mineral formation, but instead demonstrated metal-arsenic alloying at the foil edges. -

Minerals and Mineral Products in Our Bedroom Bed Hematite
Minerals and Mineral Products in our Bedroom Make-Up Kit Muscovite Bed Talc Hematite: hinges, handles, Mica mattress springs Hematite: for color Chromite: chrome plating Bismuth Radio Barite Copper: wiring Plastic Pail Quartz: clock Mica Gold: connections Cassiterite: solder Toilet Bowl / Tub Closet Feldspar: porcelain Chromite: chrome plating Pyrolusite: coloring Hematite: hinges, handles (steel) Chromite: plumbing fixtures Quartz : mirror on door Copper: tubing Desk Toothpaste Hematite: hinges, handles (steel) Apatite: teeth Chromite: chrome plating Fluorite: toothpaste Mirror Rutile: to color false Hematite: handle, frame teeth yellow Chromite: plating Gold: fillings Gold: plating Cinnabar: fillings Quartz: mirror Towels Table Lamp Sphalerite: dyes Brass (an alloy of copper and Chromite: dyes zinc): base Quartz: bulb Water Pipe/Faucet/Shower bulb Wolframite: lamp filament Brass Copper: wiring Iron Nickel Minerals and Mineral Products in our Bedroom Chrome: stainless steel Bathroom Cleaner Department of Environment and Natural Resources Borax: abrasive, cleaner, and antiseptic MINES AND GEOSCIENCES BUREAU Deodorant Spray Can Cassiterite Chromite Copper Carpet Quartz Sphalerite: dyes Telephone Chromite: dyes Drinking Glasses Copper: wiring Sulfur: foam padding Quartz Chromite: plating Gold: red color Clock Silver: electronics Pentlandite: spring Graphite: batteries Refrigerator Quartz: glass, time keeper Hematite Television Chromite: stainless steel Chromite: plating Computer Galena Wolframite: monitor Wolframite: monitor Copper Copper: -

Structure and Mineralization of Precambrian Rocks in the Galena-Roub Aix District, Black Hills, South Dakota
Structure and Mineralization of Precambrian Rocks in the \ Galena-Roubaix District, Black Hills, South Dakota ( By R. W. BAYLEY » CONTRIBUTIONS TO ECONOMIC GEOLOGY V ' GEOLOGICAL SURVEY BULLETIN 1312-E UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1970 UNITED STATES DEPARTMENT OF THE INTERIOR WALTER J. HICKEL, Secretary GEOLOGICAL SURVEY William T. Pecora, Director For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 55 cents (paper cover) CONTENTS Page Abstract_ ____-_-_----_-----__--_-----_--_--.. El Introduction _________________________________ 1 Field methods_______-_____-_____-_____-__--_ 2 General geology.___-__--__-_--__-_--___._-_--_. 3 Stratigraphy. ___________________---__--__-__-.. 4 Metabasalt and chert.______-__-__-_____-_. 4 Graphitic schist and cherty ferruginous schist. 5 Metagraywacke, schist, and slate_ ___________ 6 Structure. ___________.-______-_-__-_-_-_-___-. 6 Distribution of gold. 7 Diamond drilling. __ 9 Magnetic survey. __ 10 Summary________ 14 References. -. ______ 15 ILLUSTEATIONS Page PLATE 1. Geologic map of the Galena-Roubaix district, Lawrence County, Black Hills, South Dakota.________________ In pocket FIGURE 1. Generalized geologic map of the northern Black Hills____-___ E2 2. Magnetic survey in the vicinity of U.S. Geological Survey diamond-drill hole 1__________________________________ 13 TABLE Page TABLIS 1. Results of core-sample analyses, U.S. Geological Survey diamond-drill hole 1, Galena-Roubaix district.____________ Ell ra CONTRIBUTIONS TO ECONOMIC GEOLOGY STRUCTURE AND MINERALIZATION OF PRECAMBRIAN ROCKS IN THE GALENA-ROUB AIX DISTRICT, BLACK HILLS, SOUTH DAKOTA ByK.W.BAYLEY ABSTRACT The Galena-Roubaix district is underlain chiefly by tightly folded and mod erately metamorphosed sedimentary and volcanic rocks of Precambrian age. -

SAMPLING for COBALT at BORNITE, ALASKA Ry Jeffrey Y
SAMPLING FOR COBALT AT BORNITE, ALASKA Ry Jeffrey Y. Foley %*9 * * * * * * * * * * * * * * * * * * * * * Field Report - January, l9R 11. S. nEPARTMENT OF THE INTERIOR James S. Watt, Secretary BuREAuA OF MINES TARLE OF CONTENTS Page Introduction ................................................ Economic Geoloqy............................................ Work by the Bureau.......................................... Recommendations............................................. References ................................................. SAMDLING FOR cnRALT AT RORNITE, ALASKA By Jeffrey Y. Foley 1 INTRODIICTION Carrollite (CuCo2S4), an ore mineral of cobalt, is known to occur in the Ruby Creek Cu-Zn deposit at Bornite (fig. 1), in northwest Alaska (5, q). 2 The events leading to mineralization of dolomite and argillite units and the distribution of these rocks in the Bornite district are among the topics covered in a PhD discertation by M. W. Hitzman of Stan- ford University (in progress). Hitzman has identified carkollite and cobaltiferous pyrite at numerous intersections in diamond drill core belonging to Rear Creek Mining Corporation, the present operator of the property. A brief visit to collect bulk samples was made by a Bureau geologist in July, 19R1, as part of the Alaska Critical Metals program. ECONOMIC GEOLOGY Hitzman summarizes the distribution of cobalt as occurring: 1) "...as late cobaltiferous pyrite rims on earlier formed pyrite grains in pyritiferous, ferroan dolo- mite with disseminated sphalerite and massive siderite" 2) "...as carrollite in high-grade hornite, chalcocite, chalcopyrite, and sphalerite ore at higher levels in the deposit." 1 Geologist, U.S. Rureau of Mines, Alaska Field Operations Center, Fairbanks. 2 Underlined numbers in parentheses refer to items in the list of references at the end oF this report. ; - > . ; - .>;. -1,; g n/ /- ; > @ ! - xi #."R-: 3 2 vl- t 7:'i "^. -

Nickeline Nias C 2001-2005 Mineral Data Publishing, Version 1
Nickeline NiAs c 2001-2005 Mineral Data Publishing, version 1 Crystal Data: Hexagonal. Point Group: 6/m 2/m 2/m. Commonly in granular aggregates, reniform masses with radial structure, and reticulated and arborescent growths. Rarely as distorted, horizontally striated, {1011} terminated crystals, to 1.5 cm. Twinning: On {1011} producing fourlings; possibly on {3141}. Physical Properties: Fracture: Conchoidal. Tenacity: Brittle. Hardness = 5–5.5 VHN = n.d. D(meas.) = 7.784 D(calc.) = 7.834 Optical Properties: Opaque. Color: Pale copper-red, tarnishes gray to blackish; white with strong yellowish pink hue in reflected light. Streak: Pale brownish black. Luster: Metallic. Pleochroism: Strong; whitish, yellow-pink to pale brownish pink. Anisotropism: Very strong, pale greenish yellow to slate-gray in air. R1–R2: (400) 39.2–45.4, (420) 38.0–44.2, (440) 36.8–43.5, (460) 36.2–43.2, (480) 37.2–44.3, (500) 39.6–46.4, (520) 42.3–48.6, (540) 45.3–50.7, (560) 48.2–52.8, (580) 51.0–54.8, (600) 53.7–56.7, (620) 55.9–58.4, (640) 57.8–59.9, (660) 59.4–61.3, (680) 61.0–62.5, (700) 62.2–63.6 Cell Data: Space Group: P 63/mmc. a = 3.621(1) c = 5.042(1) Z = 2 X-ray Powder Pattern: Unknown locality. 2.66 (100), 1.961 (90), 1.811 (80), 1.071 (40), 1.328 (30), 1.033 (30), 0.821 (30) Chemistry: (1) (2) Ni 43.2 43.93 Co 0.4 Fe 0.2 As 55.9 56.07 Sb 0.1 S 0.1 Total 99.9 100.00 (1) J´achymov, Czech Republic; by electron microprobe, corresponds to (Ni0.98Co0.01 Fe0.01)Σ=1.00As1.00. -

Arsenian Sphalerite from Mina Ai,Caran, Pampa Larga
MINERALOGICAL NOTES AMERICAN MINERALOGIST, \IOL. 55, SEPTEMBER.OCTOBER, 1970 ARSENIANSPHALERITE FROM MINA AI,CARAN, PAMPALARGA, COPIAPO, CHILE Arew H. Cr-anr, Department oJ GeologicalSciences, Queen's Uniaersity, Kingston, Ontario, Canado. ABSTRACT Bright pink sphalerite from the arsenic-rich Alacr6n deposit, with a:5.4110 +0.0003 A, is shown by electron-probe microanalysis to contain only traces of iron but 17 +0.3 weight percent As INrnooucrroN The literature containsnumerous reports of appreciablesolid solution of arsenicin sphalerite(e.g. Schroll, 1953; Fleischer,1955), but the an- alytical evidenceremains largeiy unconvincing.Moss (1955;and in Hey, 1955,Frondel, 1967)proposed that the mineral voltzite has the general composition (Zn,As)S or Zn(As,S), but Frondel (1967) subsequently found no evidenceto support this suggestion.Most recent electron- probe microanalysesof sphaleritedo not record even traces of arsentc. Ilowever, Herzenberg(1932,1933) found 0.64 weight percentarsenic by chemical analysisof a raspberry-redsphalerite-like mineral from Llall- agua, Bolivia, which he named gumuc'ionite.Later workers (e.g.Hey, 1955)have assumedthat this material representsa mixture of sphalerite and realgarbut no further work appearsto have been carried out on the original or similar specimens.The presentnote describesthe occurrence of an apparently similar, and demonstrably arsenic-bearingsphalerite. Spner-Bnrrr rN rHE Ar,acnAN DBposrr Sphalerite is a minor constituent of the mineralogically complex, polymetallic Alacr6n vein deposit (Parker, Salas and P6rez, 1963),in the Pampa Larga mining district of northern Chile (Lat. 27"36'5.;Long. 70'11'W.). It occurs, however, in at least three parageneticcontexts: (1) in associationwith early arsenopyrite, pyrite, stibnite, smithite, aAsS (Clark, 1970a)and arsenolamprite(Clark, 1970b), among other minerals; (2) with late-stagegreigite, pyrite, native arsenic,realgar, and orpiment;and (3) rarely, as free-standingcrystals in vugs, in association with realgar and orpiment. -

Detection of Cds Nanoparticles and Implications for Cadmium Yellow Paint Degradation in Edvard Munch’S the Scream (C
1910 Microsc. Microanal. 23 (Suppl 1), 2017 doi:10.1017/S1431927617010212 © Microscopy Society of America 2017 Detection of CdS Nanoparticles and Implications for Cadmium Yellow Paint Degradation in Edvard Munch’s The Scream (c. 1910, Munch Museum) Barnaby D.A. Levin1, Kayla X. Nguyen1, Megan E. Holtz1, Marcie B. Wiggins2, Malcolm G. Thomas3, Eva S. Tveit4, Jennifer L. Mass5, Robert Opila6, Thomas Beebe2, David A. Muller1,7. 1. School of Applied and Engineering Physics, Cornell University, Ithaca, NY, USA. 2. Department of Chemistry and Biochemistry & UD Surface Analysis Facility, University of Delaware, Newark, DE, USA. 3. Cornell Center for Materials Research, Cornell University, Ithaca, NY, USA. 4. The Munch Museum, Tøyen, Oslo, Norway. 5. Department of Conservation, Rijksmuseum, Amsterdam, NL. 6. Department of Materials Science and Engineering, University of Delaware, Newark, DE, USA. 7. Kavli Institute for Nanoscale Science, Cornell University, Ithaca, NY, USA. Cadmium sulfide (CdS) based yellow paint is fading, flaking, and discoloring with age in billions of dollars worth of Impressionist through Expressionist masterpieces from the late 19th and early 20th centuries. Characterization of the morphology, chemistry, and crystal structure of paint particles is critical for understanding CdS pigment degradation, and the role of other cadmium compounds in paint synthesis and aging [1]. Here, we use scanning transmission electron microscopy (STEM) to identify nanoparticle structures in a sample of cadmium yellow paint from the Edvard Munch’s The Scream (c. 1910, Munch Museum), taken from a region of flaking yellow paint in the water adjacent to the two background figures on the bridge (Fig. 1a), and prepared for STEM by focused ion beam (FIB) milling. -
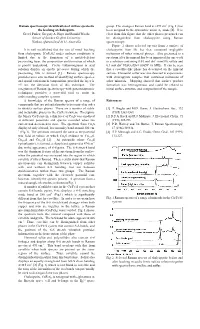
Raman Spectroscopic Identification of Surface Species in the Leaching Of
Raman spectroscopic identification of surface species in group. The strongest Raman band at »293 cm-1 (Fig. 1) has the leaching of chalcopyrite been assigned to the symmetric anion A1 mode [4]. It is Gretel Parker, Gregory A. Hope and Ronald Woods clear from this figure that the other phases presented can School of Science Griffith University be distinguished from chalcopyrite using Raman Nathan, Queensland 4111, Australia spectroscopy. Figure 2 shows selected spectra from a surface of It is well established that the rate of metal leaching chalcopyrite from Mt Isa that contained negligible from chalcopyrite [CuFeS2] under ambient conditions is inclusions of other mineral phases. Also presented is a limited due to the formation of a metal-deficient spectrum after the mineral has been immersed for one week passivating layer, the composition and formation of which in a solution containing 0.03 mol dm-3 iron(III) sulfate and -3 is poorly understood. Cyclic voltammograms in acid 0.1 mol dm H2SO4 (Eh = 0.865V vs SHE). It can be seen solution display an anodic pre-wave during which the that a covellite-like phase has developed on the mineral passivating film is formed [1]. Raman spectroscopy surface. Elemental sulfur was also detected in experiments provides an in situ method of identifying surface species, with chalcopyrite samples that contained inclusions of and spatial variations in composition, provided the layer is other minerals. Mapping showed that surface product >5 nm, the detection limit of this technique. The formation was heterogeneous and could be related to integration of Raman spectroscopy with potentiodynamic initial surface structure and composition of the sample. -

CZTS) Phases Rameez Ahmad,1 Marco Brandl,2 Monica Distaso,1 Patrick Herre,1,3 Erdmann Spiecker,3 Rainer Hock 2 and Wolfgang Peukert 1* *[email protected] 1
CrystEngComm Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/crystengcomm Page 1 of 24 CrystEngComm A comprehensive study on the mechanism behind formation and depletion of Cu 2ZnSnS 4 (CZTS) phases Rameez Ahmad,1 Marco Brandl,2 Monica Distaso,1 Patrick Herre,1,3 Erdmann Spiecker,3 Rainer Hock 2 and Wolfgang Peukert 1* *[email protected] 1. Institute of Particle Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Cauerstrasse 4, 91058 Erlangen, Germany 2. Chair for Crystallography and Structural physics, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Staudtstr. 3, 91058 Erlangen, Germany 3. Center for Nanoanalysis and Electron Microscopy (CENEM), Friedrich-Alexander-Universität Erlangen- Nürnberg (FAU), Cauerstraße 6, 91058 Erlangen, Germany Manuscript Abstract High efficiency kesterite based solar cells have vigorously raised the research interests in this material. -

The Mineralogical Fate of Arsenic During Weathering Of
THE MINERALOGICAL FATE OF ARSENIC DURING WEATHERING OF SULFIDES IN GOLD-QUARTZ VEINS: A MICROBEAM ANALYTICAL STUDY A Thesis Presented to the faculty of the Department of Geology California State University, Sacramento Submitted in partial satisfaction of the requirements for the degree of MASTER OF SCIENCE in Geology by Tamsen Leigh Burlak SPRING 2012 © 2012 Tamsen Leigh Burlak ALL RIGHTS RESERVED ii THE MINERALOGICAL FATE OF ARSENIC DURING WEATHERING OF SULFIDES IN GOLD-QUARTZ VEINS: A MICROBEAM ANALYTICAL STUDY A Thesis by Tamsen Leigh Burlak Approved by: __________________________________, Committee Chair Dr. Charles Alpers __________________________________, Second Reader Dr. Lisa Hammersley __________________________________, Third Reader Dr. Dave Evans ____________________________ Date iii Student: Tamsen Leigh Burlak I certify that this student has met the requirements for format contained in the University format manual, and that this thesis is suitable for shelving in the Library and credit is to be awarded for the project. _______________________, Graduate Coordinator ___________________ Dr. Dave Evans Date Department of Geology iv Abstract of THE MINERALOGICAL FATE OF ARSENIC DURING WEATHERING OF SULFIDES IN GOLD-QUARTZ VEINS: A MICROBEAM ANALYTICAL STUDY by Tamsen Leigh Burlak Mine waste piles within the historic gold mining site, Empire Mine State Historic Park (EMSHP) in Grass Valley, California, contain various amounts of arsenic and are the current subject of remedial investigations to characterize the arsenic present. In this study, electron microprobe, QEMSCAN (Quantitative Evaluation of Minerals by SCANning electron microscopy), and X-ray absorption spectroscopy (XAS) were used collectively to locate and identify the mineralogical composition of primary and secondary arsenic-bearing minerals at EMSHP.