Studies on Synthesis, Characterization and Applications of Microencapsulation Process Via Interfacial Polymerization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
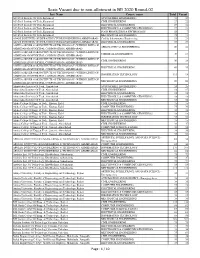
Seats Vacant Due to Non-Allotment in BE 2020 Round-02
Seats Vacant due to non-allotment in BE 2020 Round-02 Inst_Name Course_name Total Vacant A.D.Patel Institute Of Tech.,Karamsad AUTOMOBILE ENGINEERING 53 50 A.D.Patel Institute Of Tech.,Karamsad CIVIL ENGINEERING 27 22 A.D.Patel Institute Of Tech.,Karamsad ELECTRICAL ENGINEERING 27 24 A.D.Patel Institute Of Tech.,Karamsad ELECTRONICS & COMMUNICATION ENGG. 27 23 A.D.Patel Institute Of Tech.,Karamsad FOOD PROCESSING & TECHNOLOGY 53 7 A.D.Patel Institute Of Tech.,Karamsad MECHANICAL ENGINEERING 79 75 ADANI INSTITUTE OF INFRASTRUCTURE ENGINEERING,AHMEDABAD Civil & Infrastructure Engineering 53 21 ADANI INSTITUTE OF INFRASTRUCTURE ENGINEERING,AHMEDABAD ELECTRICAL ENGINEERING 53 37 ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF AERONAUTICAL ENGINEERING 90 82 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF CHEMICAL ENGINEERING 30 15 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF CIVIL ENGINEERING 30 28 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF ELECTRICAL ENGINEERING 60 55 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF INFORMATION TECHNOLOGY 111 18 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD ADITYA SILVER OAK INSTITUTE OF TECHNOLOGY (WITHIN LIMITS OF MECHANICAL ENGINEERING 90 90 AHMEDABAD MUNICIPAL CORPORATION) AHMEDABAD Ahmedabad Institute Of Tech, Ahmedabad AUTOMOBILE ENGINEERING 26 25 Ahmedabad Institute Of Tech, Ahmedabad CIVIL ENGINEERING 19 17 Ahmedabad Institute Of Tech, Ahmedabad ELECTRICAL ENGINEERING 19 18 Ahmedabad Institute Of Tech, Ahmedabad ELECTRONICS & COMMUNICATION ENGG. 19 19 Ahmedabad Institute Of Tech, Ahmedabad MECHANICAL ENGINEERING 38 36 Alpha College Of Engg. & Tech., Khatraj, Kalol CIVIL ENGINEERING 75 75 Alpha College Of Engg. -
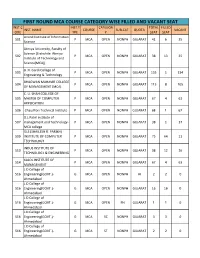
NEW WEB REPORT 24112020.Xlsx
FIRST ROUND MCA COURSE CATEGORY WISE FILLED AND VACANT SEAT INST.C INST.T CATEGOR TOTAL FILLED INST. NAME COURSE SUB-CAT. QUOTA VACANT ODE YPE Y SEAT SEAT Anand Institute of Information 501 P MCA OPEN NONPH GUJARAT 41 6 35 Science Atmiya University, Faculty of Science (Erstwhile: Atmiya 502 P MCA OPEN NONPH GUJARAT 38 13 25 Institute of Technology and Science(MCA)) B. H. Gardi College of 503 P MCA OPEN NONPH GUJARAT 135 1 134 Engineering & Technology BHAGWAN MAHAVIR COLLEGE 504 P MCA OPEN NONPH GUJARAT 113 8 105 OF MANAGEMENT (MCA) C. U. SHAH COLLEGE OF 505 MASTER OF COMPUTER P MCA OPEN NONPH GUJARAT 67 4 63 APPLICATION 506 Chaudhari Technical Institute P MCA OPEN NONPH GUJARAT 68 1 67 D.L.Patel institute of 507 management and Technology- P MCA OPEN NONPH GUJARAT 38 1 37 MCA college GLS (SHAILESH R. PARIKH) 509 INSTITUTE OF COMPUTER P MCA OPEN NONPH GUJARAT 75 64 11 TECHNOLOGY INDUS INSTITUTE OF 510 P MCA OPEN NONPH GUJARAT 38 12 26 TECHNOLOGY & ENGINEERING KALOL INSTITUTE OF 514 P MCA OPEN NONPH GUJARAT 67 4 63 MANAGEMENT L D College of 516 Engineering(GOVT.)- G MCA OPEN NONPH AI 2 2 0 Ahmedabad L D College of 516 Engineering(GOVT.)- G MCA OPEN NONPH GUJARAT 16 16 0 Ahmedabad L D College of 516 Engineering(GOVT.)- G MCA OPEN PH GUJARAT 1 1 0 Ahmedabad L D College of 516 Engineering(GOVT.)- G MCA SC NONPH GUJARAT 3 3 0 Ahmedabad L D College of 516 Engineering(GOVT.)- G MCA ST NONPH GUJARAT 2 2 0 Ahmedabad FIRST ROUND MCA COURSE CATEGORY WISE FILLED AND VACANT SEAT INST.C INST.T CATEGOR TOTAL FILLED INST. -

Admission Committee for Professional Courses Provisional Mca Institute List
ADMISSION COMMITTEE FOR PROFESSIONAL COURSES PROVISIONAL MCA INSTITUTE LIST FOR YEAR 2021-22 AS ON 30.06.2021 FEES OF ALL INSTITUTE IS AS PER 2020-21 FEES FOR YEAR 2021-22 MAY BE REVISED. YOU MAY REFER WEBSITE : www.frctech.ac.in P= SFI INSTITUTE; GOVT=GOVERNMENT;GIA=GRANT IN AID W-ONLY FEMALE INSTITUTE TOTAL SEAT SEAT TOTAL FESS OF Type of MQ NRI WITH Sr. No. Name of the Institute AFFILIATED UNIVERSITY ADDRESS OF UNIVERSITY AFTER MQ YEAR Inst. ACPC EWS 2020-21** Seats as per 2020-21* Anand Institute of Information Science (SFI)-Anand Anand Institute of Information Science GUJARAT TECHNOLOGICAL 1 P SRKSM Campus, OPP. Town Hall ,Anand - 388 001. 45 0 0 0 45 56000 (MCA) UNIVERSITY Ph. : 02692-266062, 02692-266062 Email : [email protected], web site :www.aiis.ac.in Atmiya University - Faculty of Engineering & Technology Master in Computer Application Atmiya University, Faculty of Science “YogidhamGurukul”, Kalawad Road, Rajkot-360 005. 2 (Erstwhile: Atmiya Institute of P ATMIYA UNIVERSITY 68 30 0 30 38 88000 Ph.: 0281-2563445, 0281-2563766 Technology and Science(MCA)) Mo.: 9099076159 Email : [email protected] Website:www.atmiyauni.ac.in B. H. Gardi College of Engineering & Technology (SFI)- Rajkot GardiVidyapith, Vill- Anandpar, Kalawad Road, Rajkot. B. H. Gardi College of Engineering & GUJARAT TECHNOLOGICAL 3 P Ph: 02894 – 274471-77, 09377886161, Fax : 02894 – 135 0 0 0 135 64000 Technology (MCA) UNIVERSITY 274470 Email : [email protected] web site :www.gardividyapith.ac.in Bhagwan Mahavir College of Management- (MCA ) (SFI) Vesu Surat BHAGWAN MAHAVIR COLLEGE OF BHAGWAN MAHAVIR New City Light Road, Sr No. -

Admission Committee for Professional Courses Provisional Mba Institute
ADMISSION COMMITTEE FOR PROFESSIONAL COURSES PROVISIONAL MBA INSTITUTE LIST FOR YEAR 2021-22 AS ON 30.06.2021 FEES OF ALL INSTITUTE IS AS PER 2020-21 FEES FOR YEAR 2021-22 MAY BE REVISED. YOU MAY REFER WEBSITE : www.frctech.ac.in P= SFI INSTITUTE; GOVT=GOVERNMENT; GIA=GRANT IN AID W-ONLY FEMALE INSTITUTE TOTAL TOT SEAT SEAT NR FESS OF Type of MQ AL WITH Sr. No. Name of the Institute AFFILIATED UNIVERSITY ADDRESS OF UNIVERSITY AFTER I YEAR 2020- Inst. MQ ACPC EWS 21** Seats as per 2020-21* Ahmedabad Institute of Technology Gota, Ahmedabad (SFI) Beside Vasantnagar Township Gota-Ognaj Road, off Gota Cross Road, GUJARAT TECHNOLOGICAL Ahmedabad-380 060. 1 Ahmedabad Institute of Technology P 67 30 0 30 37 71000 UNIVERSITY Ph. : (02717) 241132 / 241133 Fax : ( 02717) 241132 Email : [email protected] Website: www.aitindia.in Anand Institute of Management, Anand (SFI) GUJARAT TECHNOLOGICAL Shri R K S M Campus, Opp. Town Hall, Anand– 388 001 2 Anand Institute of Management P 67 6 0 6 61 73000 UNIVERSITY Phone / Fax : 02692269977 Email : [email protected] Website:www.aimrksm.org Faculty of Management Studies,Atmiya University Atmiya University, Faculty of Business of Master in Business Administration 3 Commerce (Erstwhile: Atmiya Institute of P ATMIYA UNIVERSITY “Yogidham Gurukul”, Kalawad Road,Rajkot-360 005. 135 60 0 60 75 88000 Technology & Science (MBA) Ph.: 0281-2563445, 0281-2563766 Email : [email protected] Website:www.atmiyauni.net Bhagwan Mahavir College of Management (SFI), Surat New City Light Road, Sr No. 149, Nr. Ashirwad Villa, B/h HeenaBunglows, BHAGWAN MAHAVIR COLLEGE OF BHAGWAN MAHAVIR 4 P BharthanaVesu, Surat. -

UNIVERSITY GRANTS COMMISSION State-Wise List of Private
UNIVERSITY GRANTS COMMISSION State-wise List of Private Universities as on 01.02.2020 S.No Name of Private University Date of Notification ARUNACHAL PRADESH 1. Apex Professional University, Pasighat, District East Siang, 10.05.2013 Arunachal Pradesh - 791102. 2. Arunachal University of Studies, NH-52, Namsai, Distt – Namsai 26.05.2012 - 792103, Arunachal Pradesh. 3. Arunodaya University, E-Sector, Nirjuli, Itanagar, Distt. Papum 21.10.2014 Pare, Arunachal Pradesh-791109 4. Himalayan University, 401, Takar Complex, Naharlagun, 03.05.2013 Itanagar, Distt – Papumpare – 791110, Arunachal Pradesh. 5. North East Frontier Technical University, Sibu-Puyi, Aalo 03.09.2014 (PO), West Siang (Distt.), Arunachal Pradesh –791001. 6. The Global University, Hollongi, Itanagar, Arunachal Pradesh. 18.09.2017 7. The Indira Gandhi Technological & Medical Sciences University, 26.05.2012 Ziro, Arunachal Pradesh. 8. Venkateshwara Open University, Itanagar, Arunachal Pradesh. 20.06.2012 S.No Andhra Pradesh 9. Bharatiya Engineering Science and Technology Innovation 17.02.2019 University, Gownivaripalli, Gorantla Mandal, Anantapur, Andhra Pradesh 10. Centurian University of Technology and Management, Gidijala 23.05.2017 Junction, Anandpuram Mandal, Visakhapatnam- 531173, Andhra Pradesh. 11. KREA University, 5655, Central, Expressway, Sri City-517646, 30.04.2018 Andhra Pradesh 12. Saveetha Amaravati University, 3rd Floor, Vaishnavi Complex, 30.04.2018 Opposite Executive Club, Vijayawada- 520008, Andhra Pradesh 13. SRM University, Neerukonda-Kuragallu Village, mangalagiri 23.05.2017 Mandal, Guntur, Dist- 522502, Andhra Pradesh (Private University) 14. VIT-AP University, Amaravati- 522237, Andhra Pradesh (Private 23.05.2017 University) ASSAM 15. Assam Don Bosco University, Azara, Guwahati 12.02.2009 16. Assam Down Town University, Sankar Madhab Path, Gandhi 29.04.2010 Nagar, Panikhaiti, Guwahati – 781 036. -

Consolidated List Private Universities
UNIVERSITY GRANTS COMMISSION State-wise List of Private Universities as on 06.08.2021 S.No Name of Private University Date of Notification ARUNACHAL PRADESH 1. Apex Professional University, Pasighat, District East Siang, 10.05.2013 Arunachal Pradesh - 791102. 2. Arunachal University of Studies, NH-52, Namsai, Distt – Namsai 26.05.2012 - 792103, Arunachal Pradesh. 3. Arunodaya University, E-Sector, Nirjuli, Itanagar, Distt. Papum 21.10.2014 Pare, Arunachal Pradesh-791109 4. Himalayan University, 401, Takar Complex, Naharlagun, 03.05.2013 Itanagar, Distt – Papumpare – 791110, Arunachal Pradesh. 5. North East Frontier Technical University, Sibu-Puyi, Aalo 03.09.2014 (PO), West Siang (Distt.), Arunachal Pradesh –791001. 6. The Global University, Hollongi, Itanagar, Arunachal Pradesh. 18.09.2017 7. The Indira Gandhi Technological & Medical Sciences University, 26.05.2012 Ziro, Arunachal Pradesh. 8. Venkateshwara Open University, Itanagar, Arunachal Pradesh. 20.06.2012 Andhra Pradesh 9. Bharatiya Engineering Science and Technology Innovation 17.02.2019 University, Gownivaripalli, Gorantla Mandal, Anantapur, Andhra Pradesh 10. Centurian University of Technology and Management, Gidijala 23.05.2017 Junction, Anandpuram Mandal, Visakhapatnam- 531173, Andhra Pradesh. 11. KREA University, 5655, Central, Expressway, Sri City-517646, 30.04.2018 Andhra Pradesh 12. Saveetha Amaravati University, 3rd Floor, Vaishnavi Complex, 30.04.2018 Opposite Executive Club, Vijayawada- 520008, Andhra Pradesh 13. SRM University, Neerukonda-Kuragallu Village, mangalagiri 23.05.2017 Mandal, Guntur, Dist- 522502, Andhra Pradesh (Private University) 14. VIT-AP University, Amaravati- 522237, Andhra Pradesh (Private 23.05.2017 University) ASSAM 15. Assam Don Bosco University, Azara, Guwahati 12.02.2009 16. Assam Down Town University, Sankar Madhab Path, Gandhi 29.04.2010 Nagar, Panikhaiti, Guwahati – 781 036. -

Name of Regional Directorate of NSS- Ahmedabad State-Gujarat
Name of Regional Directorate of NSS- Ahmedabad State-Gujarat Regional Director Name Address Email ID Telephone/Mobile/Landline Number Sh. GirdharUpadhyay Regional Directorate of NSS, [email protected] 079-26565988 2ndfloor,PatnagarYojnaBhavan, 7999894816 Ellis bridge ,Ahmedabad-380006 Secretary, dealing with NSS Name Address E Mail ID Telephone/Mobile Number Shri S.J. Haider , IAS Block No.-5, [email protected] 079-23251301/303 Principle Secretary 7th,Floor Fax 07923251325 Education Department Sachivalaya, Government of Gujarat Gandhinagar,Gujarat State NSS Officer/Officer acting as SNO Name Address E Mail ID Telephone/Mobile Number ShriYashwant Kumar HPatel Commissionerate of Higher Education [email protected] 9427685870 State NSS Officer Govt. of Gujarat, Old Sachivalaya 079-23253993 Block No.12/2,Dr. Jivraj Mehta Bhavan, Gandhinagar ,Gujarat Programme Coordinator, NSS Name Address E Mail ID Telephone/Mobile Number 1 Dr Shreedhar Nimavat Veer Narmad South Gujarat [email protected], 0261-2203039 Programme co-ordinator, NSS University, [email protected] 8780077566 Veer Narmad South Gujarat University Campus, University, Surat UdhnaMagdalla Road, Surat-395007 2 Dr. J. D. Damor Hemchandracharya [email protected] 02766-230743,Ext.316 Hemchandrcharya North North Gujarat University, 9925046204, 7573010065 Gujarat University, Patan Raj Mahal Road, P.B. No. 21 Patan-384265 3 Dr. N. K. Dobariya Saurashtra University, [email protected] 02812578501 Saurashtra University, Rajkot Kalavad Road, 9687692940 Rajkot-360 005 4 Dr. JagrutiSuvera Sardar Patel University, [email protected] 02692-226823 Sardar Patel University, VallabhVidyanagar, [email protected] 9408507810 VallabhVidyanagar,Anand Dist : Anand- 388120 5 Dr. Arunbhai Gandhi, Gujarat Vidyapith, [email protected] 079-4001630 Gujarat Vidyapeeth, Ashram Road, 9428214260 Ahmedabad Ahmedabad-380014 6 Dr. -

PROVISIONAL LIST of DEGREE ENGINEERING INSTITUTES for YEAR 2020-21 AICTE/ Management State Fees for Sr
PROVISIONAL LIST OF DEGREE ENGINEERING INSTITUTES FOR YEAR 2020-21 AICTE/ Management State Fees for Sr. College university Quota Seats Facilities Name and Address of Institute Courses NRI Intake Quota District AY-2019 No. Code INTAKE available GUJCET JEE 125% Seats (In Rs.) ## 20-21 PROVISIONAL LIST OF GOVERNMENT INSTITUTES 1 DR. S. & S.S. GHANDHY GOVERNMENT 116 CIVIL ENGINEERING 60 0 0 0 75 75 ENGINEERING COLLEGE, SURAT 116 ELECTRICAL ENGINEERING 60 0 0 0 75 75 Boys Hostel 1500/- for Ring Road, Majura Gate, Surat-395 001 116 ELECTRONICS & COMMUNICATION ENGINEERING 60 0 0 0 75 75 Surat Girls Hostel Boys Phone: 0261-2653139 Fax : 0261-2653139 116 ENVIRONMENTAL ENGINEERING 60 0 0 0 75 75 Mess Nil for Girls www.gecsu.cteguj.in 116 MECHANICAL ENGINEERING 60 0 0 0 75 75 E-mail:[email protected] 116 TOTAL 300 0 0 0 375 375 2 GOVERNMENT ENGINEERING COLLEGE, 113 CHEMICAL ENGINEERING 60 0 0 0 75 75 BHARUCH 113 CIVIL ENGINEERING 60 0 0 0 75 75 Opp. Govt. Guest House, Village Kasak, Bharuch - 113 ELECTRICAL ENGINEERING 120 0 0 0 150 150 Boys Hostel 1500/- for 392 002 113 ELECTRONICS & COMMUNICATION ENGINEERING 120 0 0 0 150 150 Bharuch Girls Hostel Boys Phone : 02642-227054 113 MECHANICAL ENGINEERING 120 0 0 0 150 150 Nil for Girls Fax : 02642-227054 www.gecbr.cteguj.in 113 TOTAL 480 0 0 0 600 600 e-mail:[email protected] 3 GOVERNMENT ENGINEERING COLLEGE, 111 CIVIL ENGINEERING 60 0 0 0 75 75 BHAVNAGAR 111 COMPUTER ENGINEERING 60 0 0 0 75 75 Near BPTI, Vidyanagar, Bhavnagar 111 ELECTRONICS & COMMUNICATION ENGINEERING 60 0 0 0 75 75 Boys Hostel 1500/- for Phone : 0278-2525354 Fax: 0278-2525354 111 INFORMATION TECHNOLOGY 60 0 0 0 75 75 Bhavnagar Girls Hostel Boys www.gecbvn.ac.in 111 MECHANICAL ENGINEERING 120 0 0 0 150 150 Mess Nil for Girls e-mail:[email protected] 111 PRODUCTION ENGINEERING 0 0 0 0 0 0 111 TOTAL 360 0 0 0 450 450 4 GOVERNMENT ENGINEERING COLLEGE, 106 CHEMICAL ENGINEERING 60 0 0 0 75 75 BHUJ, New Ravalwadi Relocation Site, Nr. -

LIST of the INSTITUTE APPROVED by NAAC/NBA in the STATE of GUJARAT in ENGINEERING/TECHNOLOGY NAAC/NBA Valid up SR
LIST OF THE INSTITUTE APPROVED BY NAAC/NBA IN THE STATE OF GUJARAT IN ENGINEERING/TECHNOLOGY NAAC/NBA Valid Up SR. NO INSTITUTES COURSES INTAKE Accredited to DHIRUBHAI AMBANI INSTITUTE OF HONS IN ICT WITH MINOR IN 66 INFORMATION AND COMMUNICATION COMPUTATIONAL SCIENCE (CS) TECHNOLOGY (University status conferred under Gujarat Act No. 6 of 2003) Nr. Indroda NAAC Valid up 1 Circle, Gandhinagar-382 007 Phone: 079- 30510574 / 575 / 576 www.daiict.ac.in ACCREDITED to 2021 INFORMATION & COMMUNICATION 264 TECHNOLOGY PANDIT DEENDAYAL PETROLEUM CHEMICAL ENGINEERING 60 UNIVERSITY*** (Affiliated to Pandit CIVIL ENGINEERING 120 Deendayal Petroleum University) Raisan ELECTRICAL ENGINEERING Village, Gandhinagar-382 007 Phone: 079- 120 COMPUTER ENGINEERING NAAC Valid up 2 23275060 Fax: 079-23275030 120 www.pdpu.ac.in INFORMATION & COMMUNICATION ACCREDITED to 2021 TECHNOLOGY 120 MECHANICAL ENGINEERING 120 PETROLEUM ENGINEERING 120 FACULTY OF TECHNOLOGY & CHEMICAL ENGINEERING 30 ENGINEERING, MSU, VADODARA CIVIL ENGINEERING 110 (Affiliated to M. S. University, Baroda M. S. COMPUTER SCIENCE & 60 University Kalabhavan, Vadodara-390 001 ENGINEERING Phone: 0265-2434188 Fax: 0265-2423898 ELECTRICAL ENGINEERING 60 www.msubaroda.ac.in ELECTRONICS ENGINEERING 30 Valid Up 3 MECHANICAL ENGINEERING 90 NAAC Accredited to 2021 METALLURGICAL AND MATERIALS 40 ENGINEERING TEXTILE ENGINEERING 30 TEXTILE PROCESSING 60 TEXTILE TECHNOLOGY 30 WATER MANAGEMENT**** 30 FACULTY OF TECHNOLOGY, DDU, CHEMICAL ENGINEERING 54 NADIAD**** (Affiliated to Dharamsinh Desai CIVIL ENGINEERING 60 University, Nadiad Dharamsinh Desai COMPUTER ENGINEERING 45 University,(DDIT), NadiadDist-Kheda-38 Valid up 4 ELECTRONICS & COMMUNICATION NAAC Accredited 7001 Phone : 0268-2520502, Fax: 0268- 45 to 2022 ENGINEERING 2520501 www.ddu.ac.in INSTRUMENTATION & CONTROL 45 ENGINEERING CHANDUBHAI S. -

Updated List of University Nodal Officers for Verification.Pdf
SCHEME OF DEVELOPING HIGH QUALITY RESEARCH LIST OF NODAL OFFICERS NAME OF NODAL SN NAME OF UNIVERSITY DEPARTMENT EMAIL-ID OFFICER AHMEDABAD UNIVERSITY Dr. DHARMESH SCHOOL OF ENGINEERING AND 1 SUBHASHRAO VARADE APPLIED SCIENCE, AHMEDABAD [email protected] UNIVERSITY ANAND AGRICULTURE DR. D. J PARMAR DEPARTMENT OF AGRICULTURAL UNIVERSITY STATISTICS, B A COLLEGE OF 2 [email protected] AGRICULTURE, ANAND ATMIYA UNIVERSITY Dr. ASHISH ADMIN OFFICE, ATMIYA UNIVERSITY 3 MAHENDRABHAI [email protected] KOTHARI AURO UNIVERSITY CHANDRASHEKHAR LIBRARIAN, AURO UNIVERSITY, VITHAL MACHANA EARTHSPACE", HAZIRA ROAD, OPP 4 [email protected] ONGC, SURAT 394510, GUJARAT, INDIA U.IN BHAGWAN MAHAVIR DR. VINEET JAIN DIRECTOR OF PHARMACY, BHAGWAN 5 UNIVERSITY MAHAVIR UNIVERSITY [email protected] BHAKT KAVI NARSINH Dr. FIROZ A SHAIKH DEPARTMENT OF ENGLISH, BHAKT 6 MEHTA UNIVERSITY KAVI NARSINH MEHTA UNIVERSITY, [email protected] JUNAGADH C U SHAH UNIVERSITY Dr. PATEL PARAGKUMAR STUDENT SCHOLARSHIP, C U SHAH 7 DINESHBHAI UNIVERSITY [email protected] CHAROTAR UNIVERSITY OF Ms. MEGHANA H. MEHTA RAMANBHAI PATEL COLLEGE OF SCIENCE AND TECHNOLOGY PHARMACY, CHAROTAR UNIVERSITY 8 [email protected] OF SCIENCE AND TECHNOLOGY NAME OF NODAL SN NAME OF UNIVERSITY DEPARTMENT EMAIL-ID OFFICER CHARUTAR VIDYA MANDAL DR. SARVESH TRIVEDI DEPARTMENT OF IT CELL, CHARUTAR 9 (CVM) VIDYA MANDAL (CVM) [email protected] CHILDREN’S UNIVERSITY PROF. (DR.) SANJAY HEAD, DEPARTMENT OF [email protected] 10 GUPTA ACCREDITATION, CHILDREN’S [email protected] UNIVERSITY, GANDHINAGAR DHARMSINH DESAI Dr. CHANDULAL FACULTY OF TECHNOLOGY, 11 UNIVERSITY KHODABHAI DHARMSINH DESAI UNIVERSITY [email protected] BHENSDADIA DR. -

List of Beneficiaries Approved Under SHODH Scheme
List of Beneficiaries approved under SHODH Scheme SN STUDENT NAME UNIVERSITY DEPARTMENT 1 GARG DIVITA DEVENDRA AHMEDABAD UNIVERSITY LIFE SCIENCES 2 GADESHA VRUNDA AHMEDABAD UNIVERSITY SCHOOL OF ENGINEERING AND KAMLESH APPLIED SCIENCE 3 JAIN STUTI PRAVEEN AHMEDABAD UNIVERSITY BIOSCIENCES AND RESEARCH LABORATORY 4 ANIYALIYA MANISHA ANAND AGRICULTURAL DEPARTMENT OF AGRICULTURAL DHIRUBHAI UNIVERSITY ENTOMOLOGY 5 BALAS DUDABHAI ANAND AGRICULTURAL SOIL AND WATER CONSERVATION BHEEKHUBHAI UNIVERSITY ENGINEERING 6 BANSAL SHAILJA ANAND AGRICULTURAL VETERINARY ANATOMY AND RAJENDRA UNIVERSITY HISTOLOGY 7 BHIMANI POOJABEN ANAND AGRICULTURAL AGRICULTURAL STATISTICS CHATURBHAI UNIVERSITY 8 BRIJESH RAJESHBHAI ANAND AGRICULTURAL DEPARTMENT OF VETERINARY HUMBAL UNIVERSITY PHARMACOLOGY AND TOXICOLOGY 9 DABHI DHARMESHKUMAR ANAND AGRICULTURAL DEPARTMENT OF HORTICULTURE MAVJIBHAI UNIVERSITY 10 DHARA RAJENDRAKUMAR ANAND AGRICULTURAL PLANT PATHOLOGY PRAJAPATI UNIVERSITY 11 DHARSHINI M S ANAND AGRICULTURAL GENETICS AND PLANT BREEDING UNIVERSITY 12 DIMPLE VASANT GOR ANAND AGRICULTURAL DEPARTMENT OF AGRICULTURAL UNIVERSITY BIOTECHNOLOGY List of Beneficiaries approved under SHODH Scheme 13 GHADIALI JATINKUMAR ANAND AGRICULTURAL PLANT PHYSIOLOGY JASHVANTBHAI UNIVERSITY 14 GUNDANIYA HIRAL ANAND AGRICULTURAL DEPARTMENT OF AGRICULTURAL VINODBHAI UNIVERSITY STATISTICS 15 KALASARIYA NEETA ANAND AGRICULTURAL DEPARTMENT OF AGRICULTURAL UKABHAI UNIVERSITY EXTENSION AND COMMUNICATION 16 MAKWANA SANJAYKUMAR ANAND AGRICULTURAL DEPARTMENT OF AGRONOMY NATVARLAL UNIVERSITY 17 -

Department of Bioscience Saurashtra University, Rajkot-360005
DEPARTMENT OF BIOSCIENCE SAURASHTRA UNIVERSITY, RAJKOT-360005 FACULTY PROFILE May - 2020 1 Name : Dr. Ramesh K. Kothari 2 Designation : Professor 3 Date of Birth : 10.01.1971 4 Address : “ISHA VASYAM”, A-6, Neel’s Green Avenue, Sanjayraj Estate, Near Saurashtra University Gate, Rajkot-360005, Gujarat-INDIA 5 E-mail ID & Web Links E-mail ID : [email protected] (Personal) [email protected] (Institutional) Webpage Link : https://sites.google.com/sauuni.ernet.in/subioscience Research Gate Link : https://www.researchgate.net/profile/Ramesh_Kothari Vidwan Database Link : https://vidwan.inflibnet.ac.in/profile/49004 Orcid Id : 0000-0003-1814-1787 Scopus Id : 8977062300 PubFacts : https://www.pubfacts.com/author_manage/addPublications/Ramesh +Kothari : Google Scholar Id https://scholar.google.co.in/citations?user=iRBdqakAAAAJ&hl=en 6 Contact Numbers Mobile : +91 9428598941 Office : +91 282 2578502-12 Ext.: 409/410 Home : +91 9428598941 7 Qualifications (Year & University) B.Sc. : Saurashtra University, Rajkot, Gujarat-INDIA M.Sc. : Saurashtra University, Rajkot, Gujarat-INDIA Saurashtra Ph.D. : University, Rajkot, Gujarat-INDIA PDF : Rutger’s University, Newark, New Jersey, United States 8 Date of Joining as Asst. Professor : 16.06.2000 Professor : 16.06.2014 9 Subjects Teaching in PG M.Sc. : Environmental Microbiology, Metagenomics, Fermentation Technology, Microbial Diversity, Extremophiles M. Phil : Metagenomics, Microbial Evolution, and Phylogeny M.Phil. / Ph.D. Coursework : Bioinformatics, Research methodology 10 Research Specializations