Substrate Enhancement Utlilizing Ribose Pretreatment in Normal and Pathologic States
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Effect of an Oral Or Intravenous Nucleotide-Free Diet on Selected Enzyme Activities in Purine Metabolism in Rats
RICE UNIVERSITY THE EFFECT OF AN ORAL OR INTRAVENOUS NUCLEOTIDE-FREE DIET ON SELECTED ENZYME ACTIVITIES IN PURINE METABOLISM IN RATS. by SANDRA LYNN SNYDER A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE MASTER OF ARTS APPROVED, THESIS COMMITTEE: .T Dr. Frederi Associate Professor of B Chemistry Dr. Susan M. B^fget Assistant Professor of Biochemistry Dr. George J. Æchfoépfer Professor of Biochemistry and Chemistry Dr. George'N. Bennett Assistant Professor of Biochemistry Dr. Roger L. Storck Professor of Biology HOUSTON, TEXAS MARCH, 1982 ABSTRACT THE EFFECT OF AN ORAL OR INTRAVENOUS NUCLEOTIDE-FREE DIET ON SELECTED ENZYME ACTIVITIES IN PURINE METABOLISM IN RATS SANDRA LYNN SNYDER Interrelationships between purine metabolism and immunity, cancer, and the diet have been considered. In studying trends of metabolic changes which occur in response to changes in the purine content of the diet, it has been hypothesized that when purines are lacking from the diet, there is a general shift from a catabolic to an anabolic state. In the present investigation, the activities of selected enzymes on purine metabolism in rats were studied with respect to an oral or intravenous nucleotide-free diet compared to the activities in rats fed normal chow. The intravenous nucleotide-free diet caused a decrease in purine nucleoside phosphorylase activity, an increase in adenine phosphoribosyl transferase, and no change in the activity of hypoxanthine-guanine phosphoribosyl transferase, with respect to a normal control diet. The orally fed nucleotide-free diet caused a decrease in the activity of purine nucleoside phosphorylase and xanthine oxidase and increase in pancreatic ribonucléase and no change in adenine phosphoribosyl transferase or hyphoxanthine-guanine phosphoribosyl transferase, with respect to a normal control diet. -

Is Human Hibernation Possible?
ANRV334-ME59-12 ARI 16 December 2007 14:50 Is Human Hibernation Possible? Cheng Chi Lee Department of Biochemistry and Molecular Biology, University of Texas Health Science Center, Houston, Texas 77030; email: [email protected] Annu. Rev. Med. 2008. 59:177–86 Key Words The Annual Review of Medicine is online at hypothermia, 5-AMP, torpor, hypometabolism http://med.annualreviews.org This article’s doi: Abstract 10.1146/annurev.med.59.061506.110403 The induction of hypometabolism in cells and organs to reduce is- Copyright c 2008 by Annual Reviews. chemia damage holds enormous clinical promise in diverse fields, in- All rights reserved cluding treatment of stroke and heart attack. However, the thought 0066-4219/08/0218-0177$20.00 that humans can undergo a severe hypometabolic state analogous to hibernation borders on science fiction. Some mammals can enter a severe hypothermic state during hibernation in which metabolic activity is extremely low, and yet full viability is restored when the animal arouses from such a state. To date, the underlying mecha- nism for hibernation or similar behaviors remains an enigma. The beneficial effect of hypothermia, which reduces cellular metabolic demands, has many well-established clinical applications. However, severe hypothermia induced by clinical drugs is extremely difficult and is associated with dramatically increased rates of cardiac arrest for nonhibernators. The recent discovery of a biomolecule, 5-AMP, which allows nonhibernating mammals to rapidly and safely enter severe hypothermia could remove this impediment and enable the wide adoption of hypothermia as a routine clinical tool. 177 ANRV334-ME59-12 ARI 16 December 2007 14:50 INTRODUCTION ing mammals. -

Enhancing Nucleotide Metabolism Protects Against Mitochondrial Dysfunction and Neurodegeneration in a PINK1 Model of Parkinson’S Disease
ARTICLES Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson's disease Roberta Tufi1, Sonia Gandhi2, Inês P. de Castro1, Susann Lehmann1, Plamena R. Angelova2, David Dinsdale1, Emma Deas2, Hélène Plun-Favreau2, Pierluigi Nicotera3, Andrey Y. Abramov2, Anne E. Willis1, Giovanna R. Mallucci1, Samantha H. Y. Loh1,4 and L. Miguel Martins1,4 Mutations in PINK1 cause early-onset Parkinson's disease (PD). Studies in Drosophila melanogaster have highlighted mitochondrial dysfunction on loss of Pink1 as a central mechanism of PD pathogenesis. Here we show that global analysis of transcriptional changes in Drosophila pink1 mutants reveals an upregulation of genes involved in nucleotide metabolism, critical for neuronal mitochondrial DNA synthesis. These key transcriptional changes were also detected in brains of PD patients harbouring PINK1 mutations. We demonstrate that genetic enhancement of the nucleotide salvage pathway in neurons of pink1 mutant flies rescues mitochondrial impairment. In addition, pharmacological approaches enhancing nucleotide pools reduce mitochondrial dysfunction caused by Pink1 deficiency. We conclude that loss of Pink1 evokes the activation of a previously unidentified metabolic reprogramming pathway to increase nucleotide pools and promote mitochondrial biogenesis. We propose that targeting strategies enhancing nucleotide synthesis pathways may reverse mitochondrial dysfunction and rescue neurodegeneration in PD and, potentially, other diseases linked to mitochondrial impairment. The role of mitochondrial impairment in PD has long been debated. By combining transcriptional and metabolic profiling, we have Recently, the identification of causative mutations in PINK1, a gene uncovered significant alterations in the nucleotide metabolism encoding a mitochondrial kinase in PD patients has renewed interest in networks of pink1 mutant flies. -

Navigating Metabolism
This is a free sample of content from Navigating Metabolism. Click here for more information on how to buy the book. Index Page references followed by b denote boxes; those followed by f denote figures; those followed by t denote tables. A Acyl-CoA synthetase, 110 Acyl transacylase, 107, 109f ACC (acetyl-CoA carboxylase), 105, 107, Adenine, 148, 149f, 155, 157f. 109f, 113, 115, 117, 173 See also Purines Acetate, 182, 193 Adenine nucleotide transporter (ANT), 38, Acetoacetate, 116b, 117b 47f, 53, 56–57 Acetone, 116b, 117b Adenosine, 142, 143f, 164–165, 164f Acetylation, 181–183, 184f Adenosine deaminase, 165 acetyl-CoA and, 9, 10f, 181–183, 184f Adenosine phosphoribosyltransferase (APRT), of lysine residues, 2, 183 155, 157f Acetyl-CoA, 2 Adenylate kinase, 16, 32, 112, 152, 154f allosteric regulation of pyruvate Adenylosuccinate, 152, 154f–155f carboxylase, 93 ADP from citrate, 43, 53, 56f, 59, 60f, 71, 72f, adenine nucleotide transporter (ANT), 38, 47f, 107, 108f 53, 56–57 citrate transporter and, 53–54, 56f adenylate kinase reaction, 16, 32, 112 entry into TCA cycle, 41, 42f energy charge, 18 in fatty acid synthesis, 107–110, 108f–109f P2 purinoreceptor activation, generation of, 39, 41, 42f, 43, 45, 45f, 164, 164f 53–54, 56f, 59 Aerobic glycolysis, 34–35, 190, 220 metabolic functions, 9, 10f, 53 Aging posttranslational modifications and, free radical theory of, 83b 181–183, 184f metabolism and, 212 Acetyl-CoA carboxylase (ACC), 105, 107, AKT, 120, 168f, 169–170, 199 109f, 113, 115, 117, 173 Alanine Acetylserotonin O-methyltransferase, β-alanine, 161–162, 163f 139, 140f conversion to pyruvate, 93 Aconitase, 60, 70 generation of, 130, 132f Activation energy, 13, 13f, 18 from pyruvate, 26 Active site, 20 urea cycle and, 134 Acyclglycerophosphate acyltransferase, 110, 111f Alanine aminotransferase, 134 ACYL (ATP-citrate lyase), 53, 56f, 59, 71, Albinism, 41 72f, 107, 108f, 182, 192 Alcohol dehydrogenase, 31f 233 © 2015 by Cold Spring Harbor Laboratory Press. -

The Effects of Acute Nicotinamide Riboside Supplementation
THE EFFECTS OF ACUTE NICOTINAMIDE RIBOSIDE SUPPLEMENTATION ON SUBSTRATE UTILISATION AND 5KM TIME-TRIAL PERFORMANCE By ELIZABETH LOUISE GRAY A thesis submitted to The University of Birmingham for the degree of MASTERS BY RESEARCH School of Sport, Exercise and Rehabilitation Sciences College of Life and Environmental Studies University of Birmingham August 2018 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Nicotinamide Riboside (NR) administration has been shown to increase fat oxidation and improve endurance performance in rodents, whilst recent research has proven it is safe for human consumption. The present study aimed to investigate the influence of acute NR supplementation on substrate utilisation and exercise performance in humans. In this counter-balanced, crossover design study, eleven recreationally-active males performed a 60-minute bout of cycling at 55% VO2max, followed by a 5km time-trial. Participants completed this twice during visits separated by at least one week, once following the consumption of 1000mg NR, and the other following placebo consumption. The contribution of fat oxidation to total substrate utilisation was not significantly different between the NR and placebo conditions during steady-state exercise (22.3±9.0% and 19.6±7.3%, respectively; p < 0.05). -

1 Targeting Subtype-Specific Metabolic Preferences in Nucleotide
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.19.049577; this version posted May 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Targeting subtype-specific metabolic preferences in nucleotide biosynthesis inhibits tumor growth in a breast cancer model Martin P. Ogrodzinski1,2, Shao Thing Teoh1, and Sophia Y. Lunt1,3* 1Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, USA 2Department of Physiology, Michigan State University, East Lansing, MI, USA 3Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, MI, USA *Correspondence: Sophia Y. Lunt, Ph.D. Biochemistry Building 603 Wilson Rd Room 522A Michigan State University East Lansing, MI 48824, USA Phone: 517-432-4886 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.19.049577; this version posted May 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Investigating metabolic rewiring in cancer can lead to the discovery of new treatment strategies for breast cancer subtypes that currently lack targeted therapies. Using MMTV- Myc driven tumors to model breast cancer heterogeneity, we investigated metabolic differences between two histological subtypes, the epithelial-mesenchymal transition (EMT) and the papillary subtypes, using a combination of genomic and metabolomic techniques. We identified differences in nucleotide metabolism between EMT and papillary subtypes: EMT tumors preferentially use the nucleotide salvage pathway, while papillary tumors prefer de novo nucleotide biosynthesis. -
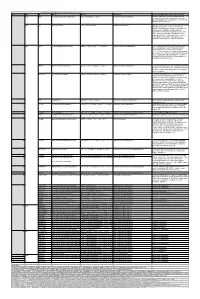
When the Reaction Is
Table S3. iJL1678-ME model modification (blocked reactions) Iter. Cat. ID Name Formula Subsystem Comments (When the reaction is turned on) 1 bp2 EDD 6-phosphogluconate dehydratase 6pgc_c⇌2ddg6p_c + h2o_c Pentose Phosphate Pathway Create a major effect of steep acetate overflow elevation in high growth. Comparing to the main glycolytic pathway, it is metabolicly less efficient but proteomicly more efficient. bp1 ICL Isocitrate lyase icit_c→glx_c + succ_c Anaplerotic Reactions Bypass for the main TCA cycle pathways from turning isocitrate to succinate, when ICL is turned on, Isocitrate dehydrogenase(ICDHyr), 2-Oxogluterate dehydrogenase(AKGDH) and Succinyl-CoA synthetase (ATP-forming,SUCOAS) would reduce. Ref. (1) and (2) shows that this reaction is off in higher growth. Ref. (3) shows that this reaction is converging to being off when the dynamic of respiration using enzyme kinetics is simulated. 2 bp1 ABTA 4-aminobutyrate transaminase 4abut_c + akg_c⇌glu__L_c + sucsal_c Arginine and Proline Metabolism Another backup pathway of succinate production, from 2-Oxoglutarate (akg). Respiration would be induced when it is on, since the flux through ETC(CYTBO3_4pp and ATPS4rpp) would increase. As it requires the co-factor pyridoxal 5'-phosphate(2−) to get catalyzed(4), indicating that this reaction is regulated by the flux of other reactions(pyridoxal 5'- phosphate(2-) production, etc.). 3 GLYAT Glycine C-acetyltransferase accoa_c + gly_c⇌2aobut_c + coa_c Glycine and Serine Metabolism A reaction that back up for the respiration. Reactions fluxes in TCA cycle would drop when this reaction is turned on. It also requires pyridoxal 5'-phosphate(2−) for the regulation. 4 NADTRHD NAD transhydrogenase nad_c + nadph_c⇌nadh_c + nadp_c Oxidative Phosphorylation A reaction that would make the transition between NAD and NADP metabolically more efficient. -

Emerging Roles of Nucleotide Metabolism in Cancer Development: Progress and Prospect
www.aging-us.com AGING 2021, Vol. 13, No. 9 Review Emerging roles of nucleotide metabolism in cancer development: progress and prospect Jingsong Ma1,2, Mengya Zhong1,2, Yubo Xiong1,2, Zhi Gao3, Zhengxin Wu4, Yu Liu5, Xuehui Hong1,2 1Institute of Gastrointestinal Oncology, School of Medicine, Xiamen University, Fujian, Xiamen 361000, China 2Department of Gastrointestinal Surgery, Zhongshan Hospital, Xiamen University, Fujian, Xiamen 361000, China 3National Center for International Research of Biological Targeting Diagnosis and Therapy, Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research, Guangxi Medical University, Guangxi, Nanning 53000, China 4Medical College of Guangxi University, Guangxi, Nanning 530000, China 5General Surgery Center, Bazhong Central Hospital, Sichuan, Bazhong 636000, China Correspondence to: Xuehui Hong; email: [email protected] Keywords: nucleotide metabolism, tumor immunity, key metabolic enzyme, signaling pathway, oncogene-induced senescence Received: August 12, 2020 Accepted: March 29, 2021 Published: May 5, 2021 Copyright: © 2021 Ma et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Abnormal cancer metabolism occurs throughout the development of tumors. Recent studies have shown that abnormal nucleotide metabolism not only accelerates the development of tumors but also inhibits the normal immune response in the tumor microenvironment. Although few relevant experiments and reports are available, study of the interaction between nucleotide metabolism and cancer development is rapidly developing. The intervention, alteration or regulation of molecular mechanisms related to abnormal nucleotide metabolism in tumor cells has become a new idea and strategy for the treatment of tumors and prevention of recurrence and metastasis. -

Dynamic Metabolic Zonation of the Hepatic Glucose Metabolism Is Accomplished by Sinusoidal Plasma Gradients of Nutrients and Hormones
ORIGINAL RESEARCH published: 12 December 2018 doi: 10.3389/fphys.2018.01786 Dynamic Metabolic Zonation of the Hepatic Glucose Metabolism Is Accomplished by Sinusoidal Plasma Gradients of Nutrients and Hormones Nikolaus Berndt 1,2 and Hermann-Georg Holzhütter 1* 1 Computational Biochemistry Group, Institute of Biochemistry, Charite—University Medicine Berlin, Berlin, Germany, 2 Institute for Computational and Imaging Science in Cardiovascular Medicine, Charite—University Medicine Berlin, Berlin, Germany Being the central metabolic organ of vertebrates, the liver possesses the largest repertoire of metabolic enzymes among all tissues and organs. Almost all metabolic pathways are resident in the parenchymal cell, hepatocyte, but the pathway capacities may largely differ depending on the localization of hepatocytes within the liver acinus-a phenomenon that is commonly referred to as metabolic zonation. Metabolic zonation is rather dynamic since gene expression patterns of metabolic enzymes may change in response to nutrition, Edited by: drugs, hormones and pathological states of the liver (e.g., fibrosis and inflammation). Steven Dooley, Universitätsmedizin Mannheim, This fact has to be ultimately taken into account in mathematical models aiming at Medizinische Fakultät Mannheim, the prediction of metabolic liver functions in different physiological and pathological Universität Heidelberg, Germany settings. Here we present a spatially resolved kinetic tissue model of hepatic glucose Reviewed by: metabolism which includes zone-specific temporal changes of enzyme abundances Adil Mardinoglu, Chalmers University of Technology, which are driven by concentration gradients of nutrients, hormones and oxygen along Sweden the hepatic sinusoids. As key modulators of enzyme expression we included oxygen, Rolf Gebhardt, Leipzig University, Germany glucose and the hormones insulin and glucagon which also control enzyme activities *Correspondence: by cAMP-dependent reversible phosphorylation. -

The Role of Sirtuin 2 Activation by Nicotinamide Phosphoribosyltransferase in the Aberrant Proliferation and Survival of Myeloid Leukemia Cells
Acute Myeloid Leukemia Articles and Brief Reports The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells Lan Dan, 1,4 Olga Klimenkova, 1 Maxim Klimiankou, 1 Jan-Henning Klusman, 2 Marry M. van den Heuvel-Eibrink, 3 Dirk Reinhardt, 2 Karl Welte, 1 and Julia Skokowa 1 1Department of Molecular Hematopoiesis, Children’s Hospital, Hannover Medical School, Hannover, Germany; 2Department of Pediatric Hematology and Oncology, Children’s Hospital, Hannover Medical School, Hannover, Germany; and 3Department of Pediatric Oncology and Hematology, Erasmus MC-Sophia Children’s Hospital, Rotterdam, The Netherlands; 4Department of Pediatrics, The First Affiliated Hospital of GuangXi Medical University, NanNing, China ABSTRACT Acknowledgments: we thank Background A. Gigina, A. Müller Brechlin Inhibitors of nicotinamide phosphoribosyltransferase have recently been validated as therapeu - and M. Reuter for their excellent tic targets in leukemia, but the mechanism of leukemogenic transformation downstream of this technical assistance. enzyme is unclear. Manuscript received on Design and Methods September 14, 2011. Revised version arrived on November 21, Here, we evaluated whether nicotinamide phosphoribosyltransferase’s effects on aberrant pro - 2011. Manuscript accepted liferation and survival of myeloid leukemic cells are dependent on sirtuin and delineated the on December 19, 2011. downstream signaling pathways operating during this process. Correspondence: Results Karl Welte, Department of We identified significant upregulation of sirtuin 2 and nicotinamide phosphoribosyltransferase Molecular Hematopoiesis, Hannover levels in primary acute myeloid leukemia blasts compared to in hematopoietic progenitor cells Medical School, Carl-Neuberg from healthy individuals. Importantly, specific inhibition of nicotinamide phosphoribosyltrans - Str. 1, 30625 Hannover, ferase or sirtuin 2 significantly reduced proliferation and induced apoptosis in human acute Germany. -

Chem331 Glycogen Metabolism
Glycogen metabolism Glycogen review - 1,4 and 1,6 α-glycosidic links ~ every 10 sugars are branched - open helix with many non-reducing ends. Effective storage of glucose Glucose storage Liver glycogen 4.0% 72 g Muscle glycogen 0.7% 245 g Blood Glucose 0.1% 10 g Large amount of water associated with glycogen - 0.5% of total weight Glycogen stored in granules in cytosol w/proteins for synthesis, degradation and control There are very different means of control of glycogen metabolism between liver and muscle Glycogen biosynthetic and degradative cycle Two different pathways - which do not share enzymes like glycolysis and gluconeogenesis glucose -> glycogen glycogenesis - biosynthetic glycogen -> glucose 1-P glycogenolysis - breakdown Evidence for two paths - Patients lacking phosphorylase can still synthesize glycogen - hormonal regulation of both directions Glycogenolysis (glycogen breakdown)- Glycogen Phosphorylase glycogen (n) + Pi -> glucose 1-p + glycogen (n-1) • Enzyme binds and cleaves glycogen into monomers at the end of the polymer (reducing ends of glycogen) • Dimmer interacting at the N-terminus. • rate limiting - controlled step in glycogen breakdown • glycogen phosphorylase - cleavage of 1,4 α glycosidic bond by Pi NOT H2O • Energy of phosphorolysis vs. hydrolysis -low standard state free energy change -transfer potential -driven by Pi concentration -Hydrolysis would require additional step s/ cost of ATP - Think of the difference between adding a phosphate group with hydrolysis • phosphorylation locks glucose in cell (imp. for muscle) • Phosphorylase binds glycogen at storage site and the catalytic site is 4 to 5 glucose residues away from the catalytic site. • Phosphorylase removes 1 residue at a time from glycogen until 4 glucose residues away on either side of 1,6 branch point – stericaly hindered by glycogen storage site • Cleaves without releasing at storage site • general acid/base catalysts • Inorganic phosphate attacks the terminal glucose residue passing through an oxonium ion intermediate. -

Glycogenosis Due to Liver and Muscle Phosphorylase Kinase Deficiency
Pediat. Res. 15: 299-303 (198 1) genetics muscle glycogenosis phosphorylase kinase deficiency liver Glycogenosis Due to Liver and Muscle Phosphorylase Kinase Deficiency N. BASHAN. T. C. IANCU. A. LERNER. D. FRASER, R. POTASHNIK. AND S. W. MOSES'"' Pediatric Research Laborarorv. Soroka Medical Center. Iaculr~of Health Sciences. Ben-Gurion Universi!,' of Negev. Beer-Sheva, and Department of Pediatrics. Carmel Hospiral. Huifa. Israel Summary hepatomegaly. The family history disclosed that two sisters were similarly affected, whereas one older brother was apparently A four-year-old Israeli Arab boy was found to have glycogen healthy. accumulation in both liver and muscle without clinical symptoms. Past history was unremarkable. The patient's height was below Liver phosphorylase kinase (PK) activity was 20% of normal, the third percentile for his age in contrast to a normal weight. He resulting in undetectable activity of phosphorylase a. Muscle PK had a doll face and a protuberant abdomen. The liver was palpable activity was about 25% of normal, resulting in a marked decrease 9 cm below the costal margin. Slight muscular hypotonia and of phosphorylase a activity. weakness were noticeable with normal tendon reflexes. He had Two sisters showed a similar pattern, whereas one brother had slightly abnormal liver function tests. a fasting blood sugar of 72 normal PK activity. The patient's liver protein kinase activity was mg %, a normal glucagon test. and no lactic acidemia or uricemia normal. Addition of exogenous protein kinase did not affect PK but slight lipidemia. Electronmicroscopic studies of a liver biopsy activity, whereas exogenous PK restored phosphorylase activity revealed marked deposition of glycogen.