DPP8/9 Inhibitors Activate the CARD8 Inflammasome in Resting Lymphocytes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DPP9 Deficiency: an Inflammasomopathy
medRxiv preprint doi: https://doi.org/10.1101/2021.01.31.21250067; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. DPP9 deficiency: an Inflammasomopathy which can be rescued by lowering NLRP1/IL-1 signaling Cassandra R. HARAPAS1,2,$, Kim S. ROBINSON3,$, Kenneth LAY4,$, Jasmine WONG4, Ricardo MORENO TRASPAS4 , Nasrin NABAVIZADEH4, Annick RAAS-ROTHSCHILD5, Bertrand BOISSON6,7,8, Scott B. DRUTMAN6, Pawat LAOHAMONTHONKUL1,2, Devon BONNER9, Mark GORRELL10, Sophia DAVIDSON1,2, Chien-Hsiung YU1,2, Hulya KAYSERILI11, Nevin HATIPOGLU12, Jean-Laurent CASANOVA6,7,8,13,14, Jonathan A. BERNSTEIN15, Franklin L. ZHONG3,16,*, Seth L. MASTERS1,2,* , Bruno REVERSADE4,10,17,* Affiliations: 1. Inflammation Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Australia 2. Department of Medical Biology, University of Melbourne, Parkville, Victoria, Australia 3. Skin Research Institute of Singapore (SRIS), A*STAR, Singapore 4. Genome Institute of Singapore (GIS), A*STAR, Singapore 5. The Institute for Rare Diseases, The Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Tel-Hashomer, Israel; Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel 6. St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, USA 7. Paris University, Imagine Institute, Paris, France NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2021.01.31.21250067; this version posted June 9, 2021. -
![DPP9 Mouse Monoclonal Antibody [Clone ID: OTI1G9] – TA504307](https://docslib.b-cdn.net/cover/2506/dpp9-mouse-monoclonal-antibody-clone-id-oti1g9-ta504307-352506.webp)
DPP9 Mouse Monoclonal Antibody [Clone ID: OTI1G9] – TA504307
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA504307 DPP9 Mouse Monoclonal Antibody [Clone ID: OTI1G9] Product data: Product Type: Primary Antibodies Clone Name: OTI1G9 Applications: FC, IHC, WB Recommended Dilution: WB 1:500~2000, IHC 1:150, FLOW 1:100 Reactivity: Human, Monkey, Mouse, Rat, Dog Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human DPP9(NP_631898) produced in HEK293T cell. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 0.7 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 96.4 kDa Gene Name: dipeptidyl peptidase 9 Database Link: NP_631898 Entrez Gene 224897 MouseEntrez Gene 485033 DogEntrez Gene 301130 RatEntrez Gene 695587 MonkeyEntrez Gene 91039 Human Q86TI2 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 DPP9 Mouse Monoclonal Antibody [Clone ID: OTI1G9] – TA504307 Background: This gene encodes a protein that is a member of the S9B family in clan SC of the serine proteases. The protein has been shown to have post-proline dipeptidyl aminopeptidase activity, cleaving Xaa-Pro dipeptides from the N-termini of proteins. -

Involvement of DPP9 in Gene Fusions in Serous Ovarian Carcinoma
Smebye et al. BMC Cancer (2017) 17:642 DOI 10.1186/s12885-017-3625-6 RESEARCH ARTICLE Open Access Involvement of DPP9 in gene fusions in serous ovarian carcinoma Marianne Lislerud Smebye1,2, Antonio Agostini1,2, Bjarne Johannessen2,3, Jim Thorsen1,2, Ben Davidson4,5, Claes Göran Tropé6, Sverre Heim1,2,5, Rolf Inge Skotheim2,3 and Francesca Micci1,2* Abstract Background: A fusion gene is a hybrid gene consisting of parts from two previously independent genes. Chromosomal rearrangements leading to gene breakage are frequent in high-grade serous ovarian carcinomas and have been reported as a common mechanism for inactivating tumor suppressor genes. However, no fusion genes have been repeatedly reported to be recurrent driver events in ovarian carcinogenesis. We combined genomic and transcriptomic information to identify novel fusion gene candidates and aberrantly expressed genes in ovarian carcinomas. Methods: Examined were 19 previously karyotyped ovarian carcinomas (18 of the serous histotype and one undifferentiated). First, karyotypic aberrations were compared to fusion gene candidates identified by RNA sequencing (RNA-seq). In addition, we used exon-level gene expression microarrays as a screening tool to identify aberrantly expressed genes possibly involved in gene fusion events, and compared the findings to the RNA-seq data. Results: We found a DPP9-PPP6R3 fusion transcript in one tumor showing a matching genomic 11;19-translocation. Another tumor had a rearrangement of DPP9 with PLIN3. Both rearrangements were associated with diminished expression of the 3′ end of DPP9 corresponding to the breakpoints identified by RNA-seq. For the exon-level expression analysis, candidate fusion partner genes were ranked according to deviating expression compared to the median of the sample set. -
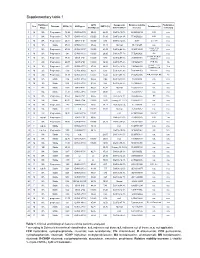
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways. -

Structural Mechanism of CARD8 Regulation by DPP9
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.13.426575; this version posted January 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Structural mechanism of CARD8 regulation by DPP9 Humayun Sharif,1,2,7 L. Robert Hollingsworth,1,2,3,7 Andrew R. Griswold,4,5,7 Jeffrey C. Hsiao,5 Qinghui Wang,6 Daniel A. Bachovchin,5,6,* and Hao Wu1,2,8,* 1Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA 2Program in Cellular and Molecular Medicine, Boston Children’s Hospital, Boston, MA 02115, USA 3Program in Biological and Biomedical Sciences, Harvard Medical School, Boston, MA 02115, USA 4Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program, New York, NY, USA 5Pharmacology Program, Weill Cornell Graduate School of Medical Sciences, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA 6Chemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA 7These authors contributed equally 8Lead Contact *Correspondence: [email protected] (H.W.), [email protected] (D.A.B.) Keywords: CARD8, NLRP1, DPP9, inflammasome, pyroptosis, Val-boroPro (VbP), cryo-EM bioRxiv preprint doi: https://doi.org/10.1101/2021.01.13.426575; this version posted January 14, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Apoptosis Ligand-Induced Enhanced Resistance to Fas/Fas
Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis This information is current as Peter Küenzi, Pascal Schneider and Dirk A. E. Dobbelaere of October 2, 2021. J Immunol 2003; 171:1224-1231; ; doi: 10.4049/jimmunol.171.3.1224 http://www.jimmunol.org/content/171/3/1224 Downloaded from References This article cites 69 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/171/3/1224.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Theileria parva-Transformed T Cells Show Enhanced Resistance to Fas/Fas Ligand-Induced Apoptosis1 Peter Ku¨enzi,2* Pascal Schneider,† and Dirk A. E. Dobbelaere3* Lymphocyte homeostasis is regulated by mechanisms that control lymphocyte proliferation and apoptosis. Activation-induced cell death is mediated by the expression of death ligands and receptors, which, when triggered, activate an apoptotic cascade. -

Genome Sequencing of Idiopathic Pulmonary Fibrosis in Conjunction with a Medical School Human Anatomy Course
Genome Sequencing of Idiopathic Pulmonary Fibrosis in Conjunction with a Medical School Human Anatomy Course Akash Kumar1,2,3, Max Dougherty1,2,3., Gregory M. Findlay1,2,3., Madeleine Geisheker1,2., Jason Klein1,2,3., John Lazar1,2., Heather Machkovech1,2,3., Jesse Resnick1,2., Rebecca Resnick1,2., Alexander I. Salter1,2., Faezeh Talebi-Liasi1., Christopher Arakawa1,2, Jacob Baudin1,2, Andrew Bogaard1,2, Rebecca Salesky1, Qian Zhou1, Kelly Smith4", John I. Clark5", Jay Shendure3", Marshall S. Horwitz4*" 1 University of Washington School of Medicine, Seattle, Washington, United States of America, 2 Medical Scientist Training Program (MSTP), University of Washington, Seattle, Washington, United States of America, 3 Department of Genome Sciences, University of Washington, Seattle, Washington, United States of America, 4 Department of Pathology, University of Washington, Seattle, Washington, United States of America, 5 Department of Biological Structure, University of Washington, Seattle, Washington, United States of America Abstract Even in cases where there is no obvious family history of disease, genome sequencing may contribute to clinical diagnosis and management. Clinical application of the genome has not yet become routine, however, in part because physicians are still learning how best to utilize such information. As an educational research exercise performed in conjunction with our medical school human anatomy course, we explored the potential utility of determining the whole genome sequence of a patient who had died following a clinical diagnosis of idiopathic pulmonary fibrosis (IPF). Medical students performed dissection and whole genome sequencing of the cadaver. Gross and microscopic findings were more consistent with the fibrosing variant of nonspecific interstitial pneumonia (NSIP), as opposed to IPF per se. -

Techniques for Immune Function Analysis Application Handbook 1St Edition
Techniques for Immune Function Analysis Application Handbook 1st Edition BD Biosciences For additional information please access the Immune Function Homepage at www.bdbiosciences.com/immune_function For Research Use Only. Not for use in diagnostic or therapeutic procedures. Purchase does not include or carry any right to resell or transfer this product either as a stand-alone product or as a component of another product. Any use of this product other than the permitted use without the express written authorization of Becton Dickinson and Company is strictly prohibited. All applications are either tested in-house or reported in the literature. See Technical Data Sheets for details. BD, BD Logo and all other trademarks are the property of Becton, Dickinson and Company. ©2003 BD Table of Contents Preface . 4 Chapter 1: Immunofluorescent Staining of Cell Surface Molecules for Flow Cytometric Analysis . 9 Chapter 2: BD™ Cytometric Bead Array (CBA) Multiplexing Assays . 35 Chapter 3: BD™ DimerX MHC:Ig Proteins for the Analysis of Antigen-specific T Cells. 51 Chapter 4: Immunofluorescent Staining of Intracellular Molecules for Flow Cytometric Analysis . 61 Chapter 5: BD FastImmune™ Cytokine Flow Cytometry. 85 Chapter 6: BD™ ELISPOT Assays for Cells That Secrete Biological Response Modifiers . 109 Chapter 7: ELISA for Specifically Measuring the Levels of Cytokines, Chemokines, Inflammatory Mediators and their Receptors . 125 Chapter 8: BD OptEIA™ ELISA Sets and Kits for Quantitation of Analytes in Serum, Plasma, and Cell Culture Supernatants. 143 Chapter 9: BrdU Staining and Multiparameter Flow Cytometric Analysis of the Cell Cycle . 155 Chapter 10: Cell-based Assays for Biological Response Modifiers . 177 Chapter 11: BD RiboQuant™ Multi-Probe RNase Protection Assay System . -

Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity
Kobe J. Med. Sci. 48, 25/31 February 2002 Molecular Genetics of Spinal Muscular Atrophy: Contribution of the NAIP Gene to Clinical Severity TOMOKO AKUTSU1*, HISAHIDE NISHIO2, KIMIAKI SUMINO2, YASUHIRO TAKESHIMA1, SYUICHI TSUNEISHI1, HIROKO WADA1, SATOSHI TAKADA3, MASAFUMI MATSUO1, 4, and HAJIME NAKAMURA1 Division of Pediatrics, Department of Development and Aging1, Division of Public Health, Department of Environmental Health and Safety2, Kobe University Graduate School of Medicine Faculty of Health Science, Kobe University School of Medicine, Kobe 654-0142, Japan3 Division of Molecular Medicine, Department of International and Environmental Medical Sciences, Kobe University Graduate School of Medicine4 Received 5 February 2002/ Accepted 19 February 2002 Key words: spinal muscular atrophy; the SMN1 gene; the NAIP gene; the p44t gene Spinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders characterized by degeneration of anterior horn cells in the spinal cord, and leads to progressive muscular weakness and atrophy. At least three SMA-related genes have been identified: SMN1, NAIP and p44t. We analyzed these genes in 32 SMA patients and found that the SMN1 gene was deleted in 30 of 32 patients (94 %), irrespective of clinical type. The NAIP gene was deleted in 6 patients and its deletion rate was higher in type I patients than that in typeⅡ or Ⅲ. Further, in type I patients lacking the NAIP gene, deterioration in their respiratory function is more rapid than in those type I patients retaining the NAIP gene. Since complete p44t deletion was observed in only 3 patients, the correlation between the p44t deletion and severity of SMA remained ambiguous. -

The Role of Nucleolin in B-Cell Lymphomas and Fas-Mediated Apoptotic Signaling
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2013 The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling Jillian F. Wise Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cancer Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Wise, Jillian F., "The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 339. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/339 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. The Role of Nucleolin in B-cell Lymphomas and Fas-Mediated Apoptotic Signaling by Jillian F Wise, BS Approved: ___________________________________________________ Felipe