Procedural Document: Rare Disease Nomenclature in English
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
SF424 Discretionary V2.1 Instructions
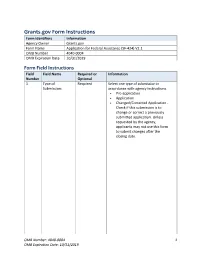
Grants.gov Form Instructions Form Identifiers Information Agency Owner Grants.gov Form Name Application for Federal Assistance (SF-424) V2.1 OMB Number 4040-0004 OMB Expiration Date 10/31/2019 Form Field Instructions Field Field Name Required or Information Number Optional 1. Type of Required Select one type of submission in Submission: accordance with agency instructions. Pre-application Application Changed/Corrected Application - Check if this submission is to change or correct a previously submitted application. Unless requested by the agency, applicants may not use this form to submit changes after the closing date. OMB Number: 4040-0004 1 OMB Expiration Date: 10/31/2019 Field Field Name Required or Information Number Optional 2. Type of Application Required Select one type of application in accordance with agency instructions. New - An application that is being submitted to an agency for the first time. Continuation - An extension for an additional funding/budget period for a project with a projected completion date. This can include renewals. Revision - Any change in the federal government's financial obligation or contingent liability from an existing obligation. If a revision, enter the appropriate letter(s). More than one may be selected. A: Increase Award B: Decrease Award C: Increase Duration D: Decrease Duration E: Other (specify) AC: Increase Award, Increase Duration AD: Increase Award, Decrease Duration BC: Decrease Award, Increase Duration BD: Decrease Award, Decrease Duration 3. Date Received: Required Enter date if form is submitted through other means as instructed by the Federal agency. The date received is completed electronically if submitted via Grants.gov. 4. -

Clinical Utility Gene Card For: 3-M Syndrome – Update 2013
European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: 3-M syndrome – Update 2013 Muriel Holder-Espinasse*,1, Melita Irving1 and Vale´rie Cormier-Daire2 European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156; published online 31 July 2013 Update to: European Journal of Human Genetics (2011) 19, doi:10.1038/ejhg.2011.32; published online 2 March 2011 1. DISEASE CHARACTERISTICS nonsense and missense mutations c.4333C4T (p.Arg1445*) and 1.1 Name of the disease (synonyms) c.4391A4C (p.His1464Pro), respectively, render CUL7 deficient 3-M syndrome (gloomy face syndrome, dolichospondylic dysplasia). in recruiting ROC1, leading to impaired ubiquitination. OBSL1: microsatellites analysis of the locus (2q35-36.1) in con- 1.2 OMIM# of the disease sanguineous families. OBSL1: microsatellites analysis of the locus 273750. (2q35-36.1) in consanguineous families. Mutations induce non- sense mediated decay. Knockdown of OBSL1 in HEK293 cells 1.3 Name of the analysed genes or DNA/chromosome segments shows the role of this gene in the maintenance of normal levels of CUL7, OBSL1 and CCDC8.1–5 CUL7. Abnormal IGFBP2 andIGFBP5 mRNA levels in two patients with OBSL1 mutations, suggesting that OBSL1 modulates the 1.4 OMIM# of the gene(s) expression of IGFBP proteins. CCDC8: microsatellites analysis 609577 (CUL7), 610991 (OBSL1) and 614145 (CCDC8). at the locus (19q13.2-q13.32). CCDC8, 1-BP DUP, 612G and CCDC8, 1-BP. -

Patient Advocacy Organizations, Industry Funding, and Conflicts of Interest
Supplementary Online Content Rose SL, Highland J, Karafa MT, Joffe S. Patient advocacy organizations, industry funding, and conflicts of interest. JAMA Intern Med. Published online January 17, 2017. doi:10.1001/jamainternmed.2016.8443 eTable 1. Search Terms Used to Identify Organizations eTable 2. Inclusion and Exclusion Criteria eTable 3. Sensitivity Analysis Comparing Survey Responses of Executive and Non-Executive Respondents of Patient Advocacy Organizations (PAOs) eFigure. Survey This supplementary material has been provided by the authors to give readers additional information about their work. © 2017 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable 1. Search Terms Used to Identify Organizations Aagenaes Syndrome Anoxia Carcinoid Aarskog Syndrome Antiphospholipid Syndrome Carcinoma Aase-Smith Syndrome II Antley-Bixler Syndrome Cardiac Abdominal Cystic Phenotype Cardiogenic Lymphangioma Anxiety Cardiogenic Shock Abdominal Obesity Metabolic Aphasia Cardiomyopathy Syndrome Apraxia Cardiovascular Achondroplasia Arrhythmia Carotid Artery Disease Achromatopsia Arteriosclerosis Celiac Acid Lipase Disease Arthritis Central Cord Syndrome Acoustic Neuroma Asperger Syndrome Cervical Cancer Acquired Hyperostosis Aspergers Chondrodysplasia Punctata Syndrome Asthma Acrocephalosyndactylia Chordoma Astrocytoma Addison Disease Churg-Strauss Syndrome Ataxia ADHD Colon Cancer Atherosclerosis Adie's syndrome Colorectal Cancer Atrial Adrenal Hyperplasia Conduct Disorder Atrial Fibulation -

Thiele's Biblical Chronology As a Corrective for Extrabiblical Dates
Andm University Seminary Studies, Autumn 1996, Vol. 34, No. 2,295-317. Copyright 1996 by Andrews University Press.. THIELE'S BIBLICAL CHRONOLOGY AS A CORRECTIVE FOR EXTRABIBLICAL DATES KENNETH A. STRAND Andrews University The outstanding work of Edwin R. Thiele in producing a coherent and internally consistent chronology for the period of the Hebrew Divided Monarchy is well known. By ascertaining and applying the principles and procedures used by the Hebrew scribes in recording the lengths of reign and synchronisms given in the OT books of Kings and Chronicles for the kings of Israel and Judah, he was able to demonstrate the accuracy of these biblical data. What has generally not been given due notice is the effect that Thiele's clarification of the Hebrew chronology of this period of history has had in furnishing a corrective for various dates in ancient Assyrian and Babylonian history. It is the purpose of this essay to look at several such dates.' 1. i%e Basic Question In a recent article in AUSS, Leslie McFall, who along with many other scholars has shown favor for Thiele's chronology, notes five vital variable factors which Thiele recognized, and then he sets forth the following opinion: In view of the complex interaction of several of the independent factors, it is clear that such factors could never have been discovered (or uncovered) if it had not been for extrabiblical evidence which established certain key absolute dates for events in Israel and Judah, such as 853, 841, 723, 701, 605, 597, and 586 B.C. It was as a result of trial and error in fitting the biblical data around these absolute dates that previous chronologists (and more recently Thiele) brought to light the factors outlined above.= '~lthou~hmuch of the information provided in this article can be found in Thiele's own published works, the presentation given here gathers it, together with certain other data, into a context and with a perspective not hitherto considered, so far as I have been able to determine. -

Eponyms in Medicine
20 Personally Speaking By Dr Cuthbert Teo Eng Swee, Editorial Board Member Eponyms in Medicine he word ‘eponym’ is derived from the Greek ‘epi’ which roughly means ‘upon’ T or ‘in addition’, and ‘onyma’ which "... in the Sciences, and means ‘name’. particularly in Medicine, the Strictly speaking, an eponym is the name of term eponym is generally the person who can be real or imaginary, from which the name of something else is derived. understood to mean Thus, Romulus is the eponym for Rome; the something (like a disease or Emperor Constantine I or Constantine the device in medicine) which Great is the eponym of Constantinople; and has been named after a Queen Victoria is the eponym for Victorian person." architecture. Some of the earliest uses of eponyms were by the ancient Greeks and ancient Romans, who named their years after their magistrates and consuls, respectively. Thus, the year 59 BC would robotics, who first introduced them in his 1942 have been known to the Greeks as Leucius, and to short story Runaround. Sometimes, eponyms are the Romans as Marcus Bibulus and Julius Caesar used to honour not the discoverer, but to honour (two consuls were elected each year). someone who is prominent in a particular field. In contemporary English, the term eponym For instance, the Hale telescope at the Palomar has also been used to refer to something which Observatory in California was not built by the is self-titled, for example, the book My Life: astronomer Gregory Hale in the 1940s, but he Bill Clinton could be described as Bill Clinton’s was instrumental in securing a grant to build Dr Teo is a eponymous book; and Gray’s Anatomy could be it. -

Trim37-Deficient Mice Recapitulate Several Features of the Multi-Organ
© 2016. Published by The Company of Biologists Ltd | Biology Open (2016) 5, 584-595 doi:10.1242/bio.016246 RESEARCH ARTICLE Trim37-deficient mice recapitulate several features of the multi- organ disorder Mulibrey nanism Kaisa M. Kettunen1,2,3,4, Riitta Karikoski5, Riikka H. Hämäläinen6, Teija T. Toivonen1, Vasily D. Antonenkov7, Natalia Kulesskaya3, Vootele Voikar3, Maarit Hölttä-Vuori8,9, Elina Ikonen8,9, Kirsi Sainio10, Anu Jalanko11, Susann Karlberg12, Niklas Karlberg12, Marita Lipsanen-Nyman12, Jorma Toppari13, Matti Jauhiainen11, J. Kalervo Hiltunen7, Hannu Jalanko14 and Anna-Elina Lehesjoki1,2,3,* ABSTRACT of the human MUL disease and thus provide a good model to study Mulibrey nanism (MUL) is a rare autosomal recessive multi-organ disease pathogenesis related to TRIM37 deficiency, including disorder characterized by severe prenatal-onset growth failure, infertility, non-alcoholic fatty liver disease, cardiomyopathy and infertility, cardiopathy, risk for tumors, fatty liver, and type 2 tumorigenesis. diabetes. MUL is caused by loss-of-function mutations in TRIM37, KEY WORDS: Mulibrey nanism, Infertility, Fatty liver, which encodes an E3 ubiquitin ligase belonging to the tripartite motif Cardiomyopathy, Tumorigenesis, Growth failure (TRIM) protein family and having both peroxisomal and nuclear localization. We describe a congenic Trim37 knock-out mouse INTRODUCTION (Trim37−/−) model for MUL. Trim37−/− mice were viable and had Mulibrey nanism (MUL) is a rare autosomal recessive multi-organ normal weight development until approximately -

Extracting and Quantifying Eponyms in Full-Text Articles Guillaume Cabanac
Extracting and quantifying eponyms in full-text articles Guillaume Cabanac To cite this version: Guillaume Cabanac. Extracting and quantifying eponyms in full-text articles. Scientometrics, Springer Verlag, 2014, vol. 98 (n° 3), pp. 1631-1645. 10.1007/s11192-013-1091-8. hal-01123700 HAL Id: hal-01123700 https://hal.archives-ouvertes.fr/hal-01123700 Submitted on 5 Mar 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Open Archive TOULOUSE Archive Ouverte (OATAO) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited version published in : http://oatao.univ-toulouse.fr/ Eprints ID : 12594 To link to this article : DOI :10.1007/s11192-013-1091-8 URL : http://dx.doi.org/10.1007/s11192-013-1091-8 To cite this version : Cabanac, Guillaume Extracting and quantifying eponyms in full-text articles. (2014) Scientometrics, vol. 98 (n° 3). pp. 1631-1645. ISSN 0138-9130 Any correspondance concerning this service should be sent to the repository administrator: [email protected] Extracting and quantifying eponyms in full-text articles Guillaume Cabanac Abstract Eponyms are known to praise leading scientists for their contributions to sci- ence. -

Repercussions of Inborn Errors of Immunity on Growth☆ Jornal De Pediatria, Vol
Jornal de Pediatria ISSN: 0021-7557 ISSN: 1678-4782 Sociedade Brasileira de Pediatria Goudouris, Ekaterini Simões; Segundo, Gesmar Rodrigues Silva; Poli, Cecilia Repercussions of inborn errors of immunity on growth☆ Jornal de Pediatria, vol. 95, no. 1, Suppl., 2019, pp. S49-S58 Sociedade Brasileira de Pediatria DOI: https://doi.org/10.1016/j.jped.2018.11.006 Available in: https://www.redalyc.org/articulo.oa?id=399759353007 How to cite Complete issue Scientific Information System Redalyc More information about this article Network of Scientific Journals from Latin America and the Caribbean, Spain and Journal's webpage in redalyc.org Portugal Project academic non-profit, developed under the open access initiative J Pediatr (Rio J). 2019;95(S1):S49---S58 www.jped.com.br REVIEW ARTICLE ଝ Repercussions of inborn errors of immunity on growth a,b,∗ c,d e Ekaterini Simões Goudouris , Gesmar Rodrigues Silva Segundo , Cecilia Poli a Universidade Federal do Rio de Janeiro (UFRJ), Faculdade de Medicina, Departamento de Pediatria, Rio de Janeiro, RJ, Brazil b Universidade Federal do Rio de Janeiro (UFRJ), Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Curso de Especializac¸ão em Alergia e Imunologia Clínica, Rio de Janeiro, RJ, Brazil c Universidade Federal de Uberlândia (UFU), Faculdade de Medicina, Departamento de Pediatria, Uberlândia, MG, Brazil d Universidade Federal de Uberlândia (UFU), Hospital das Clínicas, Programa de Residência Médica em Alergia e Imunologia Pediátrica, Uberlândia, MG, Brazil e Universidad del Desarrollo, -

Fedramp Master Acronym and Glossary Document
FedRAMP Master Acronym and Glossary Version 1.6 07/23/2020 i[email protected] fedramp.gov Master Acronyms and Glossary DOCUMENT REVISION HISTORY Date Version Page(s) Description Author 09/10/2015 1.0 All Initial issue FedRAMP PMO 04/06/2016 1.1 All Addressed minor corrections FedRAMP PMO throughout document 08/30/2016 1.2 All Added Glossary and additional FedRAMP PMO acronyms from all FedRAMP templates and documents 04/06/2017 1.2 Cover Updated FedRAMP logo FedRAMP PMO 11/10/2017 1.3 All Addressed minor corrections FedRAMP PMO throughout document 11/20/2017 1.4 All Updated to latest FedRAMP FedRAMP PMO template format 07/01/2019 1.5 All Updated Glossary and Acronyms FedRAMP PMO list to reflect current FedRAMP template and document terminology 07/01/2020 1.6 All Updated to align with terminology FedRAMP PMO found in current FedRAMP templates and documents fedramp.gov page 1 Master Acronyms and Glossary TABLE OF CONTENTS About This Document 1 Who Should Use This Document 1 How To Contact Us 1 Acronyms 1 Glossary 15 fedramp.gov page 2 Master Acronyms and Glossary About This Document This document provides a list of acronyms used in FedRAMP documents and templates, as well as a glossary. There is nothing to fill out in this document. Who Should Use This Document This document is intended to be used by individuals who use FedRAMP documents and templates. How To Contact Us Questions about FedRAMP, or this document, should be directed to [email protected]. For more information about FedRAMP, visit the website at https://www.fedramp.gov. -

2021 Western Medical Research Conference
Abstracts J Investig Med: first published as 10.1136/jim-2021-WRMC on 21 December 2020. Downloaded from Genetics I Purpose of Study Genomic sequencing has identified a growing number of genes associated with developmental brain disorders Concurrent session and revealed the overlapping genetic architecture of autism spectrum disorder (ASD) and intellectual disability (ID). Chil- 8:10 AM dren with ASD are often identified first by psychologists or neurologists and the extent of genetic testing or genetics refer- Friday, January 29, 2021 ral is variable. Applying clinical whole genome sequencing (cWGS) early in the diagnostic process has the potential for timely molecular diagnosis and to circumvent the diagnostic 1 PROSPECTIVE STUDY OF EPILEPSY IN NGLY1 odyssey. Here we report a pilot study of cWGS in a clinical DEFICIENCY cohort of young children with ASD. RJ Levy*, CH Frater, WB Galentine, MR Ruzhnikov. Stanford University School of Medicine, Methods Used Children with ASD and cognitive delays/ID Stanford, CA were referred by neurologists or psychologists at a regional healthcare organization. Medical records were used to classify 10.1136/jim-2021-WRMC.1 probands as 1) ASD/ID or 2) complex ASD (defined as 1 or more major malformations, abnormal head circumference, or Purpose of Study To refine the electroclinical phenotype of dysmorphic features). cWGS was performed using either epilepsy in NGLY1 deficiency via prospective clinical and elec- parent-child trio (n=16) or parent-child-affected sibling (multi- troencephalogram (EEG) findings in an international cohort. plex families; n=3). Variants were classified according to Methods Used We performed prospective phenotyping of 28 ACMG guidelines. -

Constrictive Pericarditis and Primary Amenorrhea with Syndactyly in an Iranian Female: Mulibrey Nanism Syndrome
TEHRAN HEART CENTER Case Report Constrictive Pericarditis and Primary Amenorrhea with Syndactyly in an Iranian Female: Mulibrey Nanism Syndrome Tahereh Davarpasand, MD, Maryam Sotoudeh Anvari, MD, Mohammad Naderan, MD*, Mohammad Ali Boroumand, MD, Hossein Ahmadi, MD Tehran Heart Center, Tehran University of Medical Sciences, Tehran, Iran. Received 25 March 2015; Accepted 10 June 2016 Abstract Mulibrey nanism is a rare autosomal recessive syndrome caused by a mutation in the TRIM37 gene with severe growth retardation and multiple organ involvement. Early diagnosis is important because 50% of the patients develop congestive heart failure owing to constrictive pericarditis, and this condition plays a critical role in the final prognosis. A 37-year-old female patient presented with symptoms of dyspnea on exertion and shortness of breath. She had severe growth failure and craniofacial dysmorphic feature. Cardiac evaluation showed constrictive pericarditis, moderate pulmonary hypertension, and mild pericardial effusion. The patient underwent pericardiectomy, but her thick and adhesive pericardium forced the surgeon to do partial pericardiotomy. Our report underlines the importance of attention to probable Mulibrey nanism when confronting patients with primary amenorrhea, growth retardation, and dysmorphic features. Early cardiac examination is of great significance in the course of the disorder, and patients must be pericardiectomized to relieve the symptoms and increase survival. J Teh Univ Heart Ctr 2016;11(4):187-191 This paper should be cited as: Davarpasand T, Sotoudeh Anvari M, Naderan M, Boroumand MA, Ahmadi H. Constrictive Pericarditis and Primary Amenorrhea with Syndactyly in an Iranian Female: Mulibrey Nanism Syndrome. J Teh Univ Heart Ctr 2016;11(4):187-191. -

Name Standards User Guide
User Guide Contents SEVIS Name Standards 1 SEVIS Name Fields 2 SEVIS Name Standards Tied to Standards for Machine-readable Passport 3 Applying the New Name Standards 3 Preparing for the New Name Standards 4 Appendix 1: Machine-readable Passport Name Standards 5 Understanding the Machine-readable Passport 5 Name Standards in the Visual Inspection Zone (VIZ) 5 Name Standards in the Machine-readable Zone (MRZ) 6 Transliteration of Names 7 Appendix 2: Comparison of Names in Standard Passports 10 Appendix 3: Exceptional Situations 12 Missing Passport MRZ 12 SEVIS Name Order 12 Unclear Name Order 14 Names-Related Resources 15 Bibliography 15 Document Revision History 15 SEVP will implement a set of standards for all nonimmigrant names entered into SEVIS. This user guide describes the new standards and their relationship to names written in passports. SEVIS Name Standards Name standards help SEVIS users: Comply with the standards governing machine-readable passports. Convert foreign names into standardized formats. Get better results when searching for names in government systems. Improve the accuracy of name matching with other government systems. Prevent the unacceptable entry of characters found in some names. SEVIS Name Standards User Guide SEVIS Name Fields SEVIS name fields will be long enough to capture the full name. Use the information entered in the Machine-Readable Zone (MRZ) of a passport as a guide when entering names in SEVIS. Field Names Standards Surname/Primary Name Surname or the primary identifier as shown in the MRZ