Molecular Signatures for Combined Targeted Treatments in Diffuse
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Malignant Peritoneal Mesothelioma: Clinical Aspects, and Therapeutic Perspectives
REVIEW ARTICLE Annals of Gastroenterology (2018) 31, 1-11 Malignant peritoneal mesothelioma: clinical aspects, and therapeutic perspectives Stergios Boussiosa, Michele Moschettab, Afroditi Karathanasia, Alexandros K. Tsiourisc, Foivos S. Kanellosc, Konstantina Tatsid, Konstantinos H. Katsanose, Dimitrios K. Christodouloue Medway NHS Foundation Trust, Kent, UK; Sarah Cannon Research Institute, London, UK; University of Ioannina, Greece; General Hospital G. Hatzikosta, Ioannina, Greece Abstract Malignant peritoneal mesothelioma (MPM) is a rare disease with a wide clinical spectrum. It arises from the peritoneal lining and commonly presents with diffuse, extensive spread throughout the abdomen and, more rarely, metastatic spread beyond the abdominal cavity. Computed tomography, magnetic resonance imaging and positron-emission tomography are important diagnostic tools used for the preoperative staging of MPM. The definitive diagnosis is based on histopathological analysis, mainly via immunohistochemistry. In this regard, paired- box gene 8 negativity represents a useful diagnostic biomarker for differentiating MPM from ovarian carcinoma. In addition, BRCA1-associated protein-1 (BAP1) loss is specific to MPM and allows it to be distinguished from both benign mesothelial lesions and ovarian serous tumors. Cytoreductive surgery (CRS) with hyperthermic intraperitoneal chemotherapy (HIPEC) has become an increasingly important therapeutic approach, while systemic therapies are still being developed. Histology, Ki-67, completeness of cytoreduction, -
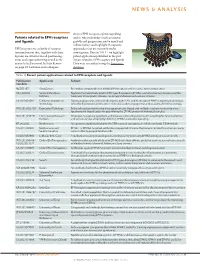
Patents Related to EPH Receptors and Ligands
NEWS & ANALYSIS discuss EPH receptor–ephrin signalling Patents related to EPH receptors and its role in disorders such as tumour and ligands growth and progression, nerve injury and inflammation, and highlight therapeutic EPH receptors are a family of receptor approaches that are currently under tyrosine kinases that, together with their investigation. Here in TABLE 1 we highlight ligands, are involved in cell positioning, patent applications published in the past tissue and organ patterning as well as the 3 years related to EPH receptors and ligands. control of cell survival. In their Review Data were researched using the Espacenet on page 39, Lackman and colleagues database. Table 1 | Recent patent applications related to EPH receptors and ligands Nature Reviews | Drug Discovery Publication Applicants Subject numbers NZ 581397 AstraZeneca Pyrimidine compounds that inhibit EPH receptors and are useful for treating cancer HK 1108702 Sanford-Burnham Peptides that selectively bind to EPH type-B receptors (EPHBs); useful for tumour imaging and the Institute treatment of neoplastic disease, neurological disease and vascular disease US 2013091591 California Institute of During angiogenesis, arterial cells express ephrin B2, and its receptor EPHB4 is expressed on venous Technology cells; this distinction can be used in methods to alter angiogenesis and to assess the effect of drugs WO 2013052710 Expression Pathology Selected reaction monitoring mass spectrometry-based and multiple reaction monitoring mass spectrometry-based assays for quantifying -

Mesothelin's Role As a Biomarker and Therapeutic Target for Malignant
cancers Review Hitting the Bull’s-Eye: Mesothelin’s Role as a Biomarker and Therapeutic Target for Malignant Pleural Mesothelioma Dannel Yeo 1,2,3 , Laura Castelletti 1,2,3 , Nico van Zandwijk 2,3,4 and John E. J. Rasko 1,2,3,5,* 1 Li Ka Shing Cell & Gene Therapy Program, The University of Sydney, Camperdown, NSW 2050, Australia; [email protected] (D.Y.); [email protected] (L.C.) 2 Faculty of Medicine and Health, The University of Sydney, Camperdown, NSW 2050, Australia; [email protected] 3 Cell and Molecular Therapies, Royal Prince Alfred Hospital, Sydney Local Health District (SLHD), Camperdown, NSW 2050, Australia 4 Concord Repatriation General Hospital, Sydney Local Health District (SLHD), Concord, NSW 2139, Australia 5 Gene and Stem Cell Therapy Program, Centenary Institute, The University of Sydney, Camperdown, NSW 2050, Australia * Correspondence: [email protected]; Tel.: +61-295656160 Simple Summary: Mesothelioma is a deadly disease with a dismal prognosis. Since its discovery, mesothelin, a cell surface protein, has been a promising biomarker and therapeutic target due to its overexpression in mesothelioma and limited expression in normal cells. This review summarizes the clinical studies that have examined mesothelin as a biomarker and therapeutic target in mesothelioma and explores future perspectives in its role to improve patient management. Abstract: Malignant pleural mesothelioma (MPM) is an aggressive cancer with limited treatment options and poor prognosis. MPM originates from the mesothelial lining of the pleura. Mesothelin Citation: Yeo, D.; Castelletti, L.; van (MSLN) is a glycoprotein expressed at low levels in normal tissues and at high levels in MPM. -

Ephb3 Suppresses Non-Small-Cell Lung Cancer Metastasis Via a PP2A/RACK1/Akt Signalling Complex
ARTICLE Received 7 Nov 2011 | Accepted 11 Jan 2012 | Published 7 Feb 2012 DOI: 10.1038/ncomms1675 EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex Guo Li1, Xiao-Dan Ji1, Hong Gao1, Jiang-Sha Zhao1, Jun-Feng Xu1, Zhi-Jian Sun1, Yue-Zhen Deng1, Shuo Shi1, Yu-Xiong Feng1, Yin-Qiu Zhu1, Tao Wang2, Jing-Jing Li1 & Dong Xie1 Eph receptors are implicated in regulating the malignant progression of cancer. Here we find that despite overexpression of EphB3 in human non-small-cell lung cancer, as reported previously, the expression of its cognate ligands, either ephrin-B1 or ephrin-B2, is significantly downregulated, leading to reduced tyrosine phosphorylation of EphB3. Forced activation of EphB3 kinase in EphB3-overexpressing non-small-cell lung cancer cells inhibits cell migratory capability in vitro as well as metastatic seeding in vivo. Furthermore, we identify a novel EphB3-binding protein, the receptor for activated C-kinase 1, which mediates the assembly of a ternary signal complex comprising protein phosphatase 2A, Akt and itself in response to EphB3 activation, leading to reduced Akt phosphorylation and subsequent inhibition of cell migration. Our study reveals a novel tumour-suppressive signalling pathway associated with kinase-activated EphB3 in non-small-cell lung cancer, and provides a potential therapeutic strategy by activating EphB3 signalling, thus inhibiting tumour metastasis. 1 Key Laboratory of Nutrition and Metabolism, Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences and Graduate School of Chinese Academy of Sciences, Shanghai 200031, China. 2 The Eastern Hepatobiliary Surgery Hospital, the Second Military Medical University, Shanghai 200433, China. -

UEMS 2020.11 Syllabus of the ETR in Rare Adult Cancers
UNION EUROPÉENNE DES MÉDECINS SPÉCIALISTES EUROPEAN UNION OF MEDICAL SPECIALISTS Association internationale sans but lucratif International non-profit organisation RUE DE L’INDUSTRIE, 24 T +32 2 649 51 64 BE- 1040 BRUSSELS F +32 2 640 37 30 www.uems.eu [email protected] UEMS 2020.11 Syllabus for residents and trainees in Rare Adult Solid Cancers The basic goal of this syllabus is to provide an understanding between the instructor and trainee so there is minimal confusion in the topics, with clear expectations. It is not a classical syllabus as it contains descriptions from different areas, but it still summarizes major and specific topics that should be covered during the training course of a resident. This syllabus is intended as supporting reference material, and the precise content and priorities of training may vary in different training institutions. The syllabus can also be modified to reflect each instructor's teaching philosophy towards the trainees. 1. There are scientific publications, web pages, and conference materials available online that could be used for educational purposes for various types of rare adult solid cancers. This is a comprehensive summary of them. 2. There are significant differences in the number of available scientific publications and reviews for different rare adult solid cancers. Some, like sarcomas, have a very robust literature, while others have been sparsely researched and consequently the availability of study materials is quite poor. 3. These differences also apply to life events and natural history. In the list of the EU CE accredited events there is a strong underrepresentation for some types of rare adult solid cancers. -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene, -

Ct Findings of Hypervascular Malignant Peritoneal Mesothelioma
Compurerized Radial. Vol. I I, No. 2, pp. 91-94, 1987 0730-4862/87 53.00 + 0.00 Printed in the U.S.A. All rights reserved Copyright 8 1987 Pergamon Journals Ltd CT FINDINGS OF HYPERVASCULAR MALIGNANT PERITONEAL MESOTHELIOMA DEBORAH S. GRANKE,* JAMES H. ELLIS and BRUCE D. RICHMOND Radiology Service (114) Veterans Administration Medical Center and Department of Radiology. University of Michigan Medical School, Ann Arbor, MI 48105. U.S.A. (Received 19 June 1986; in revised form 21 October 1986; received for publicatiorr 6 November 1986) Abstract-A case of peritoneal mesothelioma is presented in which CT demonstrated abnormal regions of increased vascularity in the omentum corresponding to hypervascular omental lesions shown by angiography. This CT appearance has not been described in prior reports of CT in peritoneal mesothelioma. Mesothelioma. peritoneal Angiography Computed tomography INTRODUCTION Reports of computed tomography (CT) in mesothelioma describe peritoneal involvement that may be extensive, with confluent tumor in layers, masses, and/or nodules and mesenteric infiltration [l, 21. A recent report of anteriography in peritoneal mesothelioma described three cases of mildly to moderately hypervascular omental lesions without arteriovenous shunting; however, the one CT scan performed was nondiagnostic [3]. We report a case of peritoneal mesothelioma where CT demon- strated abnormal regions of increased vascularity in the omentum corresponding to the hypervascular omental lesions shown by angiography. CASE REPORT A 54-year-old white male presented with a 3-month history of insidious onset of diffuse abdominal tenderness, early satiety, abdominal bloating, and crampy abdominal pain. His physical exam was unremarkable, and routine laboratory tests, sigmoidoscopy, and barium enema were normal. -

Human Ephb3 Antibody Antigen Affinity-Purified Polyclonal Sheep Igg Catalog Number: AF5667
Human EphB3 Antibody Antigen Affinity-purified Polyclonal Sheep IgG Catalog Number: AF5667 DESCRIPTION Species Reactivity Human Specificity Detects human EphB3 in direct ELISAs and Western blots. In direct ELISAs, approximately 3% crossreactivity with recombinant mouse EphB3 is observed, and less than 1% crossreactivity with recombinant rat EphB1, recombinant human (rh) EphB2 and rhEphB4 is observed. Source Polyclonal Sheep IgG Purification Antigen Affinitypurified Immunogen Mouse myeloma cell line NS0derived recombinant human EphB3 Leu38Ala550 Accession # P54753 Formulation Lyophilized from a 0.2 μm filtered solution in PBS with Trehalose. See Certificate of Analysis for details. *Small pack size (SP) is supplied either lyophilized or as a 0.2 μm filtered solution in PBS. APPLICATIONS Please Note: Optimal dilutions should be determined by each laboratory for each application. General Protocols are available in the Technical Information section on our website. Recommended Sample Concentration Western Blot 1 µg/mL See Below DATA Western Blot Detection of Human EphB3 by Western Blot. Western blot shows lysates of SHSY5Y human neuroblastoma cell line. PVDF Membrane was probed with 1 µg/mL of Sheep AntiHuman EphB3 Antigen Affinity purified Polyclonal Antibody (Catalog # AF5667) followed by HRP conjugated AntiSheep IgG Secondary Antibody (Catalog # HAF016). A specific band was detected for EphB3 at approximately 110 kDa (as indicated). This experiment was conducted under reducing conditions and using Immunoblot Buffer Group 8. PREPARATION AND STORAGE Reconstitution Sterile PBS to a final concentration of 0.2 mg/mL. Shipping The product is shipped at ambient temperature. Upon receipt, store it immediately at the temperature recommended below. -

Antitumor Activity of Mir-34A in Peritoneal Mesothelioma Relies on C-MET and AXL Inhibition
El Bezawy et al. Journal of Hematology & Oncology (2017) 10:19 DOI 10.1186/s13045-016-0387-6 RESEARCH Open Access Antitumor activity of miR-34a in peritoneal mesothelioma relies on c-MET and AXL inhibition: persistent activation of ERK and AKT signaling as a possible cytoprotective mechanism Rihan El Bezawy1, Michelandrea De Cesare1, Marzia Pennati1, Marcello Deraco2, Paolo Gandellini1, Valentina Zuco1*† and Nadia Zaffaroni1*† Abstract Background: The value of microRNAs (miRNAs) as novel targets for cancer therapy is now widely recognized. However, no information is currently available on the expression/functional role of miRNAs in diffuse malignant peritoneal mesothelioma (DMPM), a rapidly lethal disease, poorly responsive to conventional treatments, for which the development of new therapeutic strategies is urgently needed. Here, we evaluated the expression and biological effects of miR-34a—one of the most widely deregulated miRNAs in cancer and for which a lipid-formulated mimic is already clinically available—in a large cohort of DMPM clinical samples and a unique collection of in house-developed preclinical models, with the aim to assess the potential of a miR-34a-based approach for disease treatment. Methods: miR-34a expression was determined by qRT-PCR in 45 DMPM and 7 normal peritoneum specimens as well as in 5 DMPM cell lines. Following transfection with miR-34a mimic, the effects on DMPM cell phenotype, in terms of proliferative potential, apoptotic rate, invasion ability, and cell cycle distribution, were assessed. In addition, three subcutaneous and orthotopic DMPM xenograft models were used to examine the effect of miR-34a on tumorigenicity. The expression of miRNA targets and the activation status of relevant pathways were investigated by western blot. -

The Function of Human Epidermal Growth Factor Receptor-3 and Its Role in Tumors (Review)
ONCOLOGY REPORTS 30: 2563-2570, 2013 The function of human epidermal growth factor receptor-3 and its role in tumors (Review) QIN LI, ZHENYAN YUAN and BANGWEI CAO Department of Oncology, Beijing Friendship Hospital, Capital Medical University, Beijing 100050, P.R. China Received July 18, 2013; Accepted September 6, 2013 DOI: 10.3892/or.2013.2754 Abstract. Human epidermal growth factor receptor-3 (HER-3) 4 highly homologous members, HER-1 (ErbB1), HER-2 is the third member of the HER family. It was previously (ErbB2), HER-3 (ErbB3) and HER-4 (ErbB4). The distin- considered not to contain tyrosine kinase activity and catalytic guishing characteristics of the HER family are interdependent activity and the intracellular region of HER-3 could not bind and functional complementation between members. After ATP and be auto-phosphorylated. Thus, the clinical value the ligands bind to the receptor, it promotes the formation of of HER-3 was ignored. Currently, biochemical analysis has HER/ErbB receptor homodimer or heterodimer which leads to confirmed that the kinase domain of HER-3 is a specific allo- activation of the tyrosine kinase domain (1,2) and downstream steric activator; it acts as a functional activator to activate the signaling pathways (3,4). Signal transduction networks control recipient kinase (HER-1, HER-2, HER-4). With the in-depth cellular activities such as gene expression, mitosis, cell differ- knowledge of its structure and function, studies on the relation- entiation, cell proliferation, cell survival and apoptosis (1,5). ship of HER-3 and human tumors are rapidly increasing. HER-3 HER-3 is a distinctive member of the HER family as its is closely related to tumorigenesis, progression and metastasis. -

Human Ephb3 Antibody
Human EphB3 Antibody Monoclonal Mouse IgG2B Clone # 647308 Catalog Number: MAB5667 DESCRIPTION Species Reactivity Human Specificity Detects human EphB3 in direct ELISAs and Western blots. In Western blots, 100% cross-reactivity with recombinant mouse (rm) EphB3, 25% cross-reactivity with rmEphB2, and no cross-reactivity with recombinant human (rh) EphA1, A2, A5, A6, A10, rmEphA3, A4, A7, B4, B6, or recombinant rat EphB1. Source Monoclonal Mouse IgG2B Clone # 647308 Purification Protein A or G purified from hybridoma culture supernatant Immunogen Mouse myeloma cell line NS0-derived recombinant human EphB3 Leu38-Ala550 Accession # P54753 Formulation Lyophilized from a 0.2 μm filtered solution in PBS with Trehalose. See Certificate of Analysis for details. *Small pack size (-SP) is supplied either lyophilized or as a 0.2 μm filtered solution in PBS. APPLICATIONS Please Note: Optimal dilutions should be determined by each laboratory for each application. General Protocols are available in the Technical Information section on our website. Recommended Sample Concentration Western Blot 2 µg/mL See Below DATA Western Blot Detection of Human EphB3 by Western Blot. Western blot shows lysates of human cerebellum tissue. PVDF membrane was probed with 2 µg/mL of Mouse Anti-Human EphB3 Monoclonal Antibody (Catalog # MAB5667) followed by HRP-conjugated Anti-Mouse IgG Secondary Antibody (Catalog # Catalog # HAF018). A specific band was detected for EphB3 at approximately 130 kDa (as indicated). This experiment was conducted under reducing conditions and using Immunoblot Buffer Group 1. PREPARATION AND STORAGE Reconstitution Sterile PBS to a final concentration of 0.5 mg/mL. Shipping The product is shipped at ambient temperature. -

Desmoplastic Small Round Cell Tumor of the Abdomen: a Case Report and Literature Review of Therapeutic Options
Vol.4, No.4, 207-211 (2012) Health http://dx.doi.org/10.4236/health.2012.44031 Desmoplastic small round cell tumor of the abdomen: A case report and literature review of therapeutic options Hafida Benhammane1*, Leila Chbani2, Abdelmalek Ousadden3, Ouadii Mouquit3, Siham Tizniti4, Afaf Riffi Amarti2, Nouafal Mellas1, Omar El Mesbahi1 1Department of Medical Oncology, Hassan II University Hospital, Fez, Morocco; *Corresponding Author: [email protected] 2Department of Pathology, Hassan II University Hospital, Fez, Morocco 3Department of General Surgery, Hassan II University Hospital, Fez, Morocco 4Department of Radiology, Hassan II University Hospital, Fez, Morocco Received 22 December 2011; revised 18 January 2012; accepted 6 February 2012 ABSTRACT rent therapeutic options include multiagent chemothe- rapy and aggressive surgical debulking and radiotherapy Desmoplastic small round cell tumor (DSRCT) is [4,5]. The addition of hyperthermic intra-peritoneal che- a rare and highly aggressive variety of sarcoma motherapy in the multimodal approach has been reported arising typically from abdominal or pelvic peri- in very few cases but no effect on survival has been clearly toneum. Diagnosis and treatment approaches of demonstrated [6]. this entity are complex and require a skilled, ex- The prognosis of this disease is poor with a Median perienced, multidisciplinary team. Authors re- survival of 17 months approximately [7]. port their experience with a case of an intra-ab- We report a case of an intra-abdominal DSRCT in a 37 dominal DSRCT arising in a 37-year-old young -year-old young man who was treated with combination man in order to discuss the clinico-pathological chemotherapy and surgery.