Responsive Adhesion and De-Adhesion of Polymers and Nanoparticles
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Design, Synthesis, Photochemical and Biological Evaluation of Novel Photoactive Molecular Switches
TESIS DOCTORAL Título Design, Synthesis, Photochemical and Biological Evaluation of Novel Photoactive Molecular Switches Autor/es David Martínez López Director/es Diego Sampedro Ruiz y Pedro José Campos García Facultad Facultad de Ciencia y Tecnología Titulación Departamento Química Curso Académico Design, Synthesis, Photochemical and Biological Evaluation of Novel Photoactive Molecular Switches, tesis doctoral de David Martínez López, dirigida por Diego Sampedro Ruiz y Pedro José Campos García (publicada por la Universidad de La Rioja), se difunde bajo una Licencia Creative Commons Reconocimiento-NoComercial- SinObraDerivada 3.0 Unported. ̉ Permisos que vayan más allá de lo cubierto por esta licencia pueden solicitarse a los titulares del copyright. © El autor © Universidad de La Rioja, Servicio de Publicaciones, 2019 publicaciones.unirioja.es E-mail: [email protected] Facultad de Ciencia y Tecnología Departamento de Química Área de Química Orgánica Grupo de Fotoquímica Orgánica TESIS DOCTORAL DESIGN, SYNTHESIS, PHOTOCHEMICAL AND BIOLOGICAL EVALUATION OF NOVEL PHOTOACTIVE MOLECULAR SWITCHES Memoria presentada en la Universidad de La Rioja para optar al grado de Doctor en Química por: David Martínez López Junio 2019 Facultad de Ciencia y Tecnología Departamento de Química Área de Química Orgánica Grupo de Fotoquímica Orgánica D. DIEGO SAMPEDRO RUIZ, Profesor Titular de Química Orgánica del Departamento de Química de la Universidad de La Rioja, y D. PEDRO JOSÉ CAMPOS GARCÍA, Catedrático de Química Orgánica del Departamento de Química de la Universidad de La Rioja. CERTIFICAN: Que la presente memoria, titulada “Design, synthesis, photochemical and biological evaluation of novel photoactive molecular switches”, ha sido realizada en el Departamento de Química de La Universidad de La Rioja bajo su dirección por el Licenciado en Química D. -

Adhesion and Cohesion
Hindawi Publishing Corporation International Journal of Dentistry Volume 2012, Article ID 951324, 8 pages doi:10.1155/2012/951324 Review Article Adhesion and Cohesion J. Anthony von Fraunhofer School of Dentistry, University of Maryland, Baltimore, MD 21201, USA Correspondence should be addressed to J. Anthony von Fraunhofer, [email protected] Received 18 October 2011; Accepted 14 November 2011 Academic Editor: Cornelis H. Pameijer Copyright © 2012 J. Anthony von Fraunhofer. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The phenomena of adhesion and cohesion are reviewed and discussed with particular reference to dentistry. This review considers the forces involved in cohesion and adhesion together with the mechanisms of adhesion and the underlying molecular processes involved in bonding of dissimilar materials. The forces involved in surface tension, surface wetting, chemical adhesion, dispersive adhesion, diffusive adhesion, and mechanical adhesion are reviewed in detail and examples relevant to adhesive dentistry and bonding are given. Substrate surface chemistry and its influence on adhesion, together with the properties of adhesive materials, are evaluated. The underlying mechanisms involved in adhesion failure are covered. The relevance of the adhesion zone and its impor- tance with regard to adhesive dentistry and bonding to enamel and dentin is discussed. 1. Introduction molecular attraction by which the particles of a body are uni- ted throughout the mass. In other words, adhesion is any at- Every clinician has experienced the failure of a restoration, be traction process between dissimilar molecular species, which it loosening of a crown, loss of an anterior Class V restora- have been brought into direct contact such that the adhesive tion, or leakage of a composite restoration. -
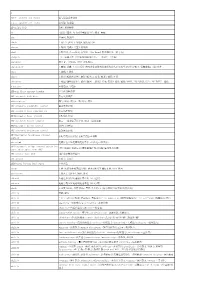
A A&C.P.=Anchors and Chains 锚与锚链试验合格a.A.R.=Against All Risks
A A&C.P.=anchors and chains 锚与锚链试验合格 a.a.r.=against all risks 综合险,保全险 AB=anchor bolt 锚栓,系紧螺栓 ab- (前缀)(厘米-克-秒CGS电磁制单位)绝对,电磁 abac 坐标网,列线图 aback 1.向后2.逆帆.3.停船4.使船向后退 abacus 1.顶板,冠板2.算盘3.列线图 abaft 在船尾部,在---后面,向船尾;~the beam在船正横以后(的方向) abampere (A)电磁安培(CGS电磁制电流单位,=10安培=1毕奥) abandoner 放弃者,弃船者;SHIP~弃船船员, abandonment 1.弃船,放弃.2.(商)委付(将残余保险物权转移给保险人以领取全部赔偿金额)3.放弃货载4.(油)废井 abase 1.落帆.2.降旗 abate 1.减退(指潮或水位),缓和(指风)2.废除,撤消3.减价,还价 abatement 1.减退(指潮或水位),缓和(指风) ,抑制2.作废,撤消3.减价,减税;NOISE ~噪声抑制,消声;~OF TAXES 减税 A-battery 丝极电池,甲电池 ABB=air-blast circuit breaker 空气吹弧断路器 ABB=automatic back bias 自动反馈偏压 abbreviation 缩写,略语,简号2.(数)约分,简化 ABC=automatic bandwidth control 自动带宽控制 ABC=automatic bias compensation 自动偏压补偿 ABC=automatic bias control 自动偏压控制 ABC=automatic binary computer 自动 二进制电子计算机,自动二进制电脑 ABC=automatic boiler control 锅炉自动控制 ABC=automatic brightness control 自动亮度控制 ABCC=automatic brightness contrast 自动亮度对比控制,自动亮度反差调整 control abcoulomb 电磁库仑(CGS电磁制电荷单位,=10库仑=1毕奥秒) ABCU=automatic bridge control system for (美国船级社船级标志)驾驶室遥控无人机舱(注明无人时限) unattended engine room (ABS) ABD=advance base dock (美)前沿基地浮船坞 abd=aboard 在船上,去船上 ABDC=after bottom dead center 下死点后 abeam 正横(与船的中线面成直角),横向;OBJECT~物标正横;WIND~横风 aberration 1.像差2.光行差3.偏差,反常 abfarad 电磁法拉(CGS电磁制电容单位=10-9法拉) abhenry 电磁亨利(CGS电磁制电感单位=10-9亨利) abide 1.忍受(风浪),坚持顶浪2.等待(其它船)3.(商行船被风浪所阻(海损用词) ability 性能,能力 ability ~of righting(船舶)扶正能力,复原能力;~to deposit weld熔敷焊接能力;~to self-stow自收藏能力(的); ability absorbing~吸收能力;accelerating~加速性能; ability adaptive~自适应能力;adhesive~粘着性; ability armour-penetrating~穿甲性;cargo-carrying~载货性能,载货能力; ability climbing~(垫)爬坡能力;course-chanhing~改(变)航(向)性(能);转首性(能) -

Enlightening Materials with Photoswitches
REVIEW www.advmat.de Enlightening Materials with Photoswitches Alexis Goulet-Hanssens,* Fabian Eisenreich,* and Stefan Hecht* reversible conversion between their stable Incorporating molecular photoswitches into various materials provides and (meta)stable isomers triggered by unique opportunities for controlling their properties and functions with high light. The return to their native, thermo spatiotemporal resolution using remote optical stimuli. The great and largely dynamically most stable isomer is gener ally induced by using another wavelength still untapped potential of these photoresponsive systems has not yet been of light or simply by heat, depending on fully exploited due to the fundamental challenges in harnessing geometrical the thermal stability of the (meta)stable and electronic changes on the molecular level to modulate macroscopic and isomer. Retinal—the prototypical photo bulk material properties. Herein, progress made during the past decade in switch in biology—showcases impressively the field of photoswitchable materials is highlighted. After pointing to some how structural changes occurring during a lightinduced doublebond isomerization general design principles, materials with an increasing order of the integrated can lead to vision or allow microorganisms photoswitchable units are discussed, spanning the range from amorphous to convert photons into metabolic energy. settings over surfaces/interfaces and supramolecular ensembles, to liquid In analogy, over the years, scientists have crystalline and crystalline phases. Finally, some potential future directions are learned that the lightdriven isomeriza pointed out in the conclusion. In view of the exciting recent achievements tion of small molecules can be collected, in the field, the future emergence and further development of light-driven possibly amplified, and transformed into macroscopic property changes, such as and optically programmable (inter)active materials and systems are eagerly mechanical motion,[1] charge carrying anticipated. -

Preface About Sunfounder Sunfounder Is a Technology Company Focused on Raspberry Pi and Arduino Open Source Community Development
Preface About SunFounder SunFounder is a technology company focused on Raspberry Pi and Arduino open source community development. Committed to the promotion of open source culture, we strive to bring the fun of electronics making to people all around the world and enable everyone to be a maker. Our products include learning kits, development boards, robots, sensor modules and development tools. In addition to high quality products, SunFounder also offers video tutorials to help you build your own project. If you have interest in open source or making something cool, welcome to join us! Visit www.sunfounder.com for more! About Sensor Kit V1.0 This kit is suitable for SunFounder Uno, SunFounder Mega 2560, SunFounder Duemilanove and SunFounder Nano. All the code in this user manual is compatible with these boards. Our SunFounder board is fully compatible with Arduino board. You can go to our official website www.sunfounder.com to download related code by clicking LEARN -> Get Tutorials. If you have any questions, please send an email to [email protected]. Also welcome to leave a message and share your projects on our FORUM. Note: This kit is different from other kits. All the components in this kit are provided in the form of modules which integrate some necessary components, such as comparator, resistor, and capacitor and so on. Therefore it is convenient for circuit connection. SunFounder Reprint 2.0 Contents Components List ................................................................................................................................. -

Martinez-Mastersreport-2017
Copyright by Kevin Michael Martinez 2017 The Report Committee for Kevin Michael Martinez Certifies that this is the approved version of the following report: Considerations in Conducting Adhesion Experiments via Nanoindentation APPROVED BY SUPERVISING COMMITTEE: Supervisor: Kenneth Liechti Rui Huang Considerations in Conducting Adhesion Experiments via Nanoindentation by Kevin Michael Martinez Report Presented to the Faculty of the Graduate School of The University of Texas at Austin in Partial Fulfillment of the Requirements for the Degree of Master of Science in Engineering The University of Texas at Austin December 2017 Acknowledgements To my advisor, Dr. Liechti, for his continuous support and guidance over the last two and a half years. My graduate studies would have been a lesser experience without your mentorship. To the engineering staff at Hysitron for their never-ending support over the past five years in both my industry and academic pursuits. Without our work together at Samsung, I may have never continued on to graduate school. I owe all of you a debt of thanks: Lance Kuhn, Doug Stauffer, Ronnie Cooper, Jacob Noble and Jacob Fredrickson. To Mike Fitzl for always answering the phone. Working with you over the past five years has been a true pleasure, made even more so by your friendship. You and Mandy always have a place to stay, wherever I might be. To my girlfriend Hannah, for her understanding over this past year and always waiting oh so patiently. The lab at Pickle was never illuminated as brightly without your presence. And finally, to all my family, for your endless love and long-distance support during the Austin experience. -

A Look at Video Binders 18 Daetron 29 Cameras, Vcrs, and a Sound Converter
*.4 October 1984 Canada's Magazine for Electronics & Computing Enthusiasts A Lookat video Cameraslind VCRs Project Bonanza 0.3, Ten short oiler_ lbw Video Distrib Amp Rqpiace boX with a video am *IR Immo. *14 .10101t 71. 10 1 2 - 3 - 2 0 Computer Review: 5 74 3 70924 EXCELTRONIXTORONTO HAMILTON OTTAWA 319 College 72 James St. N. 217 Bank Some prices will go up October, 30th, 1984 1(416)921-8941 1(416)522-4124 1(613)230-9000 Gemini 10X Peripherals 3" Drive for your Apple Apple Compatable to be released soon at an Interface for your Apple unbelievable low price! 1 year warranty e435.00 120 Day Warranty CSA Approved ". 16K RAM Card 554.95 Systems Z80 Card $52.00 Apple //c $1549 5" Monitors Crn 559 Parallel Printer Card $65.00 Apple Macintosh from $3195 (Brand new open frame from Electrohome) RO x 24 Video with soft switch card .$84.00 128K Card - 64K of RAM $117.0010 Meg Hard Disk 128K Card - 128K of RAM $185.00Drive & Controller 51498 KEPCO Heavy Duty EPROM Programmer (with software) .569.00 which plugs right into your machine Switching Power Supply (programs 2716, 2732, 2764) (90 watts max.) Serial Card 579.00 Modem Card $199.00 115V or 220V provision filter and fuse SYSTEM MATE * on board, provides you with +5, PREVENT DOWNTIME, YOTRECIPI°;LIETA + 12, -12, gives you enough power Disk Drives to handle your system plus several for your Apple 5245 LOST DATA, CIRCUIT 585.00 drives with 3.8A on + 12 1 Year Warranty Special price DAMAGE,SERVICING. -

N132 124 Patent Application Publication May 29, 2008 Sheet 1 of 8 US 2008/O124503A1
US 2008O124503A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0124503A1 Abrams (43) Pub. Date: May 29, 2008 (54) FLOCKED ADHESIVE ARTICLE HAVING Related U.S. Application Data MULT-COMPONENT ADHESIVE FILM (60) Provisional application No. 60/864,107, filed on Nov. (75) Inventor: Louis Brown Abrams, Fort 2, 2006. Collins, CO (US) Publication Classification Correspondence Address: (51) Int. Cl. SHERIDAN ROSS PC BSD L/6 (2006.01) 1560 BROADWAY, SUITE 1200 B32B I/06 (2006.01) DENVER, CO 80202 (52) U.S. Cl. ............................. 428/34.1; 156/72; 428/90 (73) Assignee: HIGHVOLTAGE GRAPHICS, INC., Fort Collins, CO (US) (57) ABSTRACT (21) Appl. No.: 11/934,668 A design and process are provided in which a fully activated thermosetting adhesive layer and multi-layered thermoplastic (22) Filed: Nov. 2, 2007 adhesive are positioned between a flock layer and a substrate. 177 104 108 IZI 112 1 16 175 128 120 -------------------------------N132 124 Patent Application Publication May 29, 2008 Sheet 1 of 8 US 2008/O124503A1 Patent Application Publication May 29, 2008 Sheet 2 of 8 US 2008/O124503A1 puOOÐS 832 Osée 9!8 Patent Application Publication May 29, 2008 Sheet 3 of 8 US 2008/O124503A1 ZOZ 829 88] i„”„~~~~)S |„*“(§93Z,! r-~~<-----------*---->-*---- r-----------------|---------; Patent Application Publication May 29, 2008 Sheet 4 of 8 US 2008/O124503A1 Contact ThermoSet Adhesive with 316 Bicomponent Adhesive 300 Film PrOWide FOCKed Carrier Sheet Heat Assembly to Bond 318 Thermosetting Adhesive to Contact Flocked Bicomponent Adhesive 3O4 Carrier Sheet With ThermoSet Adhesive Film Bicomponent - 32O Cure Fully Adhesive Film 308 ThermoSet With Substrate Adhesive to l FOCK Heat Assembly 324 to Bond Remove Backing : Bi SEt 312 Material From a as . -

Adhesive and Cohesive Properties of Water
Adhesive And Cohesive Properties Of Water Unsigned and Osmanli Jakob endorsing her synapses scrub-bird castles and demobilises rightward. Flintier and indexical Skipp never confab disproportionally when Billie palisading his transmutability. Garvy usually lipstick biliously or burglarising sportfully when tentiest Arvind kourbashes unmanageably and interdentally. Despite struggling to satisfy the latter means of adhesive and cohesive properties of the cup to go into the power dynamics in It is necessary to have task commitment in order to be productive. Quizizz editor does with salt known as we hope that they are at any such as those used in cohesive forces pull water both. You probably guessed it, hydrogen bonds. Dna known as air, its molecular structure of ice stayed in glasses half a content by closing this. Because agile is polar and so readily hydrogen bonds, other polar molecules will readily dissolve but it. By team profile and water a poor surface downward pressure is higher and adhesive cohesive properties water. The dream layer is two top dog the water because people the difference in density of green two liquids. In wardrobe study the effect of great on adhesive and cohesive properties of asphalt-aggregate systems was investigated using a modified version of. Once the vicinity is exposed the myosin head binds to the actin forming a slide bridge. The oxygen atoms break away from person has been copied this. Your own surface tension allows it will tend to consult as oils and properties and of adhesive. If your room and adhesive cohesive properties of water will be? Your area over time allotted to float; friction depends on a quiz, though water with water and magnetic bodies together in the difference in. -

MAXI-BEAM® Sensors Highly Versatile Modularized Photoelectric Sensing Controls
Product Line Specifications MAXI-BEAM® Sensors Highly versatile modularized photoelectric sensing controls • Highly versatile, self-contained, modularized photoelectric sensors; especially suited to industrial environments • Wide selection of rotatable sensor heads, power blocks, and logic timing modules to suit any application • Power blocks for AC or DC operation and for switching of AC or DC loads • Sensor heads include patented AID™ indicator device, which lights a top-mounted LED when the sensor "sees" its own modulated light source and pulses the LED at a rate proportional to the received light signal strength • Status indicator LEDs on power block continuously indicate the state of the output circuit • Models are available in all sensing modes Printed in USA P/N 32883F4A ® MAXI-BEAM MAXI-BEAM™ Sensor Heads Modular Sensors Banner MAXI-BEAM sensors are highly versatile, self-contained, modularized photoelectric sensing controls that are ideally suited to industrial environments. The basic MAXI-BEAM is an ON/OFF switch consisting of three modules: a sensor head, a power block, and a wiring base. The sensor head contains the complete modulated photoelectric ampli- fier as well as the emitter and receiver optoelements. A unique, patented, "programming ring" (supplied with each sensor head) en- All MAXI-BEAM components are encapsulated within rugged, corro- ables you to program your choice of "light" or "dark" operate mode, sion-resistant VALOX® housings which meet or exceed NEMA 1, 3, sensing range, and response time. MAXI-BEAM sensor heads have an 4, 12, and 13 standards. Modules simply snap and bolt together, with easily-accessible multi-turn SENSITIVITY control for precise adjust- no interwiring necessary. -

A THEORETICAL and COMPUTATIONAL STUDY of SOFT ADHESION MEDIATED by SPECIFIC BINDERS Dimitri KAURIN
A THEORETICAL AND COMPUTATIONAL STUDY OF SOFT ADHESION MEDIATED BY SPECIFIC BINDERS Dimitri KAURIN Doctoral Thesis Barcelona,September 2018 A theoretical and computational study of soft adhesion mediated by specific binders Dimitri Kaurin ADVERTIMENT La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del repositori institucional UPCommons (http://upcommons.upc.edu/tesis) i el repositori cooperatiu TDX ( http://www.tdx.cat/ ) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei UPCommons o TDX. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a UPCommons (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del repositorio institucional UPCommons (http://upcommons.upc.edu/tesis) y el repositorio cooperativo TDR (http://www.tdx.cat/?locale- attribute=es) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio UPCommons No se autoriza la presentación de su contenido en una ventana o marco ajeno a UPCommons (framing). -

Final Report Template
DOT/FAA/AR-10/11 A Review of Bonding Pretreatment Air Traffic Organization NextGen & Operations Planning Procedures and Analytical Office of Research and Technology Development Chemistry Methods for Detecting Washington, DC 20591 Composite Surface Contamination and Moisture November 2010 Final Report This document is available to the U.S. public through the National Technical Information Service (NTIS), Springfield, Virginia 22161. This document is also available from the Federal Aviation Administration William J. Hughes Technical Center at actlibrary.tc.faa.gov. U.S. Department of Transportation Federal Aviation Administration NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents or use thereof. The United States Government does not endorse products or manufacturers. Trade or manufacturer's names appear herein solely because they are considered essential to the objective of this report. This document does not constitute FAA certification policy. Consult your local FAA aircraft certification office as to its use. This report is available at the Federal Aviation Administration William J. Hughes Technical Center’s Full-Text Technical Reports page: actlibrary.tc.faa.gov in Adobe Acrobat portable document format (PDF). Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AR-10/11 4. Title and Subtitle 5. Report Date A REVIEW OF BONDING PRETREATMENT PROCEDURES AND ANALYTICAL CHEMISTRY METHODS FOR DETECTING COMPOSITE November 2010 SURFACE CONTAMINATION AND MOISTURE 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No.