New Angiogenic Regulators Produced by Tams: Perspective for Targeting Tumor Angiogenesis
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
Peripheral Mononuclear Blood Cells Contribute to The
Genes Nutr (2015) 10:11 DOI 10.1007/s12263-015-0460-8 RESEARCH PAPER Peripheral mononuclear blood cells contribute to the obesity- associated inflammatory state independently of glycemic status: involvement of the novel proinflammatory adipokines chemerin, chitinase-3-like protein 1, lipocalin-2 and osteopontin 1,2,3 1,2,3 1,2,3 Victoria Catala´n • Javier Go´mez-Ambrosi • Amaia Rodrı´guez • 1,2,3 2,4 3,5 2,3,6 Beatriz Ramı´rez • Vı´ctor Valentı´ • Rafael Moncada • Camilo Silva • 2,6 1,2,3,6 Javier Salvador • Gema Fru¨hbeck Received: 24 December 2014 / Accepted: 28 March 2015 / Published online: 14 April 2015 Ó Springer-Verlag Berlin Heidelberg 2015 Abstract Inflammation is a critical contributor to the including chemerin, chitinase-3-like protein 1 (YKL-40), pathogenesis of metabolic disorders with adipose tissue lipocalin-2 (LCN-2) and osteopontin (OPN), and their being crucial in the inflammatory response by releasing circulating concentrations were also determined by ELISA. multiple adipokines with either pro- or anti-inflammatory We show, for the first time, that PBMC gene expression activities with potential functions as metabolic regulators. levels of chemerin (P \ 0.0001), chitinase-3-like protein 1 Peripheral blood mononuclear cells (PBMC) have been (P = 0.010), lipocalin-2 (P \ 0.0001) and osteopontin proposed as representative of the inflammatory status in (P \ 0.0001) were strongly upregulated in obesity inde- obesity. The aim of the present study was to evaluate the pendently of the glycemic state. Circulating concentrations contribution of PBMC to the obesity-associated chronic of these adipokines followed the same trend being sig- inflammation analyzing the expression of novel adipokines. -

Semaphorin7a Promotes Tumor Growth and Exerts a Pro-Angiogenic Effect in Macrophages of Mammary Tumor-Bearing Mice
ORIGINAL RESEARCH ARTICLE published: 05 February 2014 doi: 10.3389/fphys.2014.00017 Semaphorin7A promotes tumor growth and exerts a pro-angiogenic effect in macrophages of mammary tumor-bearing mice Ramon Garcia-Areas 1, Stephania Libreros 1, Samantha Amat 1, Patricia Keating 2, Roberto Carrio 3, Phillip Robinson 4, Clifford Blieden 5 and Vijaya Iragavarapu-Charyulu 1* 1 Tumor Immunology, Department of Biomedical Sciences, Florida Atlantic University, Boca Raton, FL, USA 2 Immunology, Department of Biological Sciences, Florida Atlantic University, Boca Raton, FL, USA 3 Tumor Immunology, Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL, USA 4 Department of Clinical Sciences, Florida Atlantic University, Boca Raton, FL, USA 5 Department of Pathology and Laboratory Medicine, Jackson Memorial Hospital, University of Miami Miller School of Medicine, Miami, FL, USA Edited by: Semaphorins are a large family of molecules involved in axonal guidance during the Michal A. Rahat, Technion - Israel development of the nervous system and have been recently shown to have both Institute for Technology, Israel angiogenic and anti-angiogenic properties. Specifically, semaphorin 7A (SEMA7A) has Reviewed by: been reported to have a chemotactic activity in neurogenesis and to be an immune Andrea Foskett, Texas A&M Health α β Science Center, USA modulator through 1 1integrins. SEMA7A has been shown to promote monocyte Jincai Luo, The University of Tokyo, chemotaxis and induce them to produce proinflammatory mediators. In this study we Japan explored the role of SEMA7A in a murine model of breast cancer. We show that SEMA7A Zahava Vadasz, Bnai-Zion Medical is highly expressed by DA-3 murine mammary tumor cells in comparison to normal Center, Israel mammary cells (EpH4), and that peritoneal elicited macrophages from mammary tumor- *Correspondence: Vijaya Iragavarapu-Charyulu, Tumor bearing mice also express SEMA7A at higher levels compared to those derived from Immunology, Department of normal mice. -

Genome-Wide Analysis Identifies Critical DNA Methylations Within
Chen et al. J Transl Med (2021) 19:73 https://doi.org/10.1186/s12967-021-02740-6 Journal of Translational Medicine RESEARCH Open Access Genome-wide analysis identifes critical DNA methylations within NTRKs genes in colorectal cancer Zijian Chen1,3† , Zenghong Huang2,3†, Yanxin Luo2,3, Qi Zou3,4, Liangliang Bai3, Guannan Tang3, Xiaolin Wang3, Guangwen Cao5, Meijin Huang2,3, Jun Xiang1* and Huichuan Yu3* Abstract Background: Neurotrophic tropomyosin receptor kinases (NTRKs) are a gene family function as oncogene or tumor suppressor gene in distinct cancers. We aimed to investigate the methylation and expression profles and prognostic value of NTRKs gene in colorectal cancer (CRC). Methods: An analysis of DNA methylation and expression profles in CRC patients was performed to explore the critical methylations within NTRKs genes. The methylation marker was validated in a retrospectively collected cohort of 229 CRC patients and tested in other tumor types from TCGA. DNA methylation status was determined by quantita- tive methylation-specifc PCR (QMSP). Results: The profles in six CRC cohorts showed that NTRKs gene promoter was more frequently methylated in CRC compared to normal mucosa, which was associated with suppressed gene expression. We identifed a specifc methylated region within NTRK3 promoter targeted by cg27034819 and cg11525479 that best predicted survival outcome in CRC. NTRK3 promoter methylation showed independently predictive value for survival outcome in the validation cohort (P 0.004, HR 2.688, 95% CI [1.355, 5.333]). Based on this, a nomogram predicting survival outcome was developed with= a C-index of 0.705. Furthermore, the addition of NTRK3 promoter methylation improved the performance of currently-used prognostic model (AIC: 516.49 vs 513.91; LR: 39.06 vs 43.64, P 0.032). -

Primary Antibodies Flyer
Primary Antibodies Your choice of size and format Format Concentration Size CF® dye conjugates (13 colors) 0.1 mg/mL 100 or 500 uL Biotin, HRP or AP conjugates 0.1 mg/mL 100 or 500 uL R-PE, APC, or Per-CP conjugates 0.1 mg/mL 250 uL Purified, with BSA 0.1 mg/mL 100 or 500 uL Purified, BSA-free (Mix-n-Stain™ Ready) 1 mg/mL 50 uL Advantages Figure 1. IHC staining of human prostate Figure 2. Flow cytometry analysis of U937 • More than 1000 monoclonal antibodies carcinoma with anti-ODC1 clone cells with anti-CD31/PECAM clone C31.7, • Growing selection of monoclonal rabbit ODC1/485. CF647 conjugate (blue) or isotype control (orange). antibodies • Validated in IHC and other applications Your choice of 13 bright and photostable CF® dyes • Choose from 13 bright and stable CF® dyes CF® dye Ex/Em (nm) Features • Also available with R-PE, APC, PerCP, HRP, AP, CF®405S 404/431 • Better fit for the 450/50 flow cytometer channel than Alexa Fluor® 405 or biotin CF®405M 408/452 • More photostable than Pacific Blue®, with less green spill-over • Purified antibodies available BSA-free, 1 mg/mL, • Compatible with super-resolution imaging by SIM ready to use for Mix-n-Stain™ labeling or other CF®488A 490/515 • Less non-specific binding and spill-over than Alexa Fluor® 488 conjugation • Very photostable and pH-insensitive • Compatible with super-resolution imaging by TIRF • Offered in affordable 100 uL size CF®543 541/560 • Brighter than Alexa Fluor® 546 CF®555 555/565 • Brighter than Cy®3 • Validated in multicolor super-resolution imaging by STORM CF®568 -

S100 Calcium-Binding Protein S100 Proteins
S S100 Calcium-Binding Protein experiments showed the S100 protein fraction consti- tuted two different dimeric species comprised of two ▶ S100 Proteins b protomers (S100B) or an a, b heterodimer (Isobe et al. 1977). Early members of the S100 protein family were frequently given suffixes based on their localiza- tion or molecular size and included S100P (placental), S100 Proteins S100C (cardiac or calgizzarin), p11 (11 kDa), and MRP8/MRP14 (myeloid regulatory proteins, 8 and Brian R. Dempsey, Anne C. Rintala-Dempsey and 14 kDa). In 1993, initial genetic studies showed that Gary S. Shaw six of the S100 genes were clustered on chromosome Department of Biochemistry, The University of 1q21 (Engelkamp et al. 1993), a number that has Western Ontario, London, ON, Canada expanded since. Based on this observation most of the proteins were renamed according to the physical order they occupy on the chromosome. These include Synonyms S100A1 (formerly S100a), S100A2 (formerly S100L), S100A10 (p11), S100A8/S100A14 (MRP8/MRP14). S100 calcium-binding protein A few S100 proteins are found on other chromosomes including S100B (21q21). Currently there are 27 known S100 family members: S100A1-A18, S100B, S100 Protein Family Members S100G, S100P, S100Z, trichohylin, filaggrin, filaggrin- 2, cornulin, and repetin (Table 1). S100A1, S100A2, S100A3, S100A4, S100A5, S100A6, S100A7, S100A8, S100A9, S100A10, S100A11, S100A12, S100A13, S100A14, S100A15, S100A16, Role of S100 Proteins in Calcium Signaling S100B, S100P, S100G, S100Z, trichohylin, filaggrin, filaggrin-2, -

Review Article S100 Protein Family in Human Cancer
Am J Cancer Res 2014;4(2):89-115 www.ajcr.us /ISSN:2156-6976/ajcr0000257 Review Article S100 protein family in human cancer Hongyan Chen, Chengshan Xu, Qing’e Jin, Zhihua Liu The State Key Laboratory of Molecular Oncology, Cancer Institute and Hospital, Chinese Academy of Medical Sci- ences and Peking Union Medical College, Beijing 100021, China Received January 16, 2014; Accepted February 10, 2014; Epub March 1, 2014; Published March 15, 2014 Abstract: S100 protein family has been implicated in multiple stages of tumorigenesis and progression. Among the S100 genes, 22 are clustered at chromosome locus 1q21, a region frequently rearranged in cancers. S100 protein possesses a wide range of intracellular and extracellular functions such as regulation of calcium homeostasis, cell proliferation, apoptosis, cell invasion and motility, cytoskeleton interactions, protein phosphorylation, regulation of transcriptional factors, autoimmunity, chemotaxis, inflammation and pluripotency. Many lines of evidence suggest that altered expression of S100 proteins was associated with tumor progression and prognosis. Therefore, S100 proteins might also represent potential tumor biomarkers and therapeutic targets. In this review, we summarize the evidence connecting S100 protein family and cancer and discuss the mechanisms by which S100 exerts its diverse functions. Keywords: S100 proteins, proliferation, apoptosis, invasion, migration, pluripotency, biomarker Introduction and their relationship with different cancers because of their involvement in a variety of bio- The S100 gene family is the largest subfamily logical events which are closely related to of calcium binding proteins of EF-hand type [1]. tumorigenesis and cancer progression. The To date, at least 25 distinct members of this association between S100 proteins and cancer subgroup have been described. -

Supporting Online Material
1 2 3 4 5 6 7 Supplementary Information for 8 9 Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic 10 retinopathy 11 12 *Samuel A. Mills, *Andrew I. Jobling, *Michael A. Dixon, Bang V. Bui, Kirstan A. Vessey, Joanna A. Phipps, 13 Ursula Greferath, Gene Venables, Vickie H.Y. Wong, Connie H.Y. Wong, Zheng He, Flora Hui, James C. 14 Young, Josh Tonc, Elena Ivanova, Botir T. Sagdullaev, Erica L. Fletcher 15 * Joint first authors 16 17 Corresponding author: 18 Prof. Erica L. Fletcher. Department of Anatomy & Neuroscience. The University of Melbourne, Grattan St, 19 Parkville 3010, Victoria, Australia. 20 Email: [email protected] ; Tel: +61-3-8344-3218; Fax: +61-3-9347-5219 21 22 This PDF file includes: 23 24 Supplementary text 25 Figures S1 to S10 26 Tables S1 to S7 27 Legends for Movies S1 to S2 28 SI References 29 30 Other supplementary materials for this manuscript include the following: 31 32 Movies S1 to S2 33 34 35 36 1 1 Supplementary Information Text 2 Materials and Methods 3 Microglial process movement on retinal vessels 4 Dark agouti rats were anaesthetized, injected intraperitoneally with rhodamine B (Sigma-Aldrich) to label blood 5 vessels and retinal explants established as described in the main text. Retinal microglia were labelled with Iba-1 6 and imaging performed on an inverted confocal microscope (Leica SP5). Baseline images were taken for 10 7 minutes, followed by the addition of PBS (10 minutes) and then either fractalkine or fractalkine + candesartan 8 (10 minutes) using concentrations outlined in the main text. -

Calprotectin and Calgranulin C Serum Levels in Bacterial Sepsis
Diagnostic Microbiology and Infectious Disease 93 (2019) 219–226 Contents lists available at ScienceDirect Diagnostic Microbiology and Infectious Disease journal homepage: www.elsevier.com/locate/diagmicrobio ☆ Calprotectin and calgranulin C serum levels in bacterial sepsis Eva Bartáková a,MarekŠtefan a,Alžběta Stráníková a, Lenka Pospíšilová b, Simona Arientová a,Ondřej Beran a, Marie Blahutová b, Jan Máca a,c,MichalHoluba,⁎ a Department of Infectious Diseases, First Faculty of Medicine, Charles University and Military University Hospital Prague, U Vojenské nemocnice 1200, 169 02 Praha 6, Czech Republic b Department of Clinical Biochemistry, Military University Hospital Prague, U Vojenské nemocnice 1200, 169 02 Praha 6, Czech Republic c Department of Anesthesiology and Intensive Care Medicine, University Hospital of Ostrava, 17. listopadu 1790/5, 708 52 Ostrava-Poruba, Czech Republic article info abstract Article history: The aim of this study was to evaluate the serum levels of calprotectin and calgranulin C and routine biomarkers in Received 26 March 2018 patients with bacterial sepsis (BS). The initial serum concentrations of calprotectin and calgranulin C were signif- Received in revised form 2 October 2018 icantly higher in patients with BS (n = 66) than in those with viral infections (n = 24) and the healthy controls Accepted 10 October 2018 (n = 26); the level of calprotectin was found to be the best predictor of BS, followed by the neutrophil- Available online 17 October 2018 lymphocyte count ratio (NLCR) and the level of procalcitonin (PCT). The white blood cell (WBC) count and the NLCR rapidly returned to normal levels, whereas PCT levels normalized later and the increased levels of Keywords: Sepsis calprotectin, calgranulin C, and C-reactive protein persisted until the end of follow-up. -

Proteomic Analysis of Two Non-Bronchoscopic Methods of Sampling the Lungs of Patients with the Acute Respiratory Distress Syndrome (ARDS)
Clin Proteom (2007) 3:30–41 DOI 10.1007/s12014-007-9002-8 Proteomic Analysis of Two Non-Bronchoscopic Methods of Sampling the Lungs of Patients with the Acute Respiratory Distress Syndrome (ARDS) Dong W. Chang & Giuseppe Colucci & Tomas Vaisar & Trevor King & Shinichi Hayashi & Gustavo Matute-Bello & Roger Bumgarner & Jay Heinecke & Thomas R. Martin & Guido M. Domenighetti Published online: 5 January 2008 # Humana Press Inc. 2007 Abstract BAL samples, 13.2% were increased in s-Cath compared to Objective The collection of lung fluid using a suction catheter mini-BAL, and 18.4% were decreased in s-Cath compared (s-Cath) and non-bronchoscopic bronchoalveolar lavage to mini-BAL. For each of the seven subjects, overabun- (mini-BAL) are two minimally invasive methods of sampling dance analysis showed that the actual number of differen- the distal airspaces in patients with the acute respiratory tially expressed spots in the mini-BAL and s-Cath sample distress syndrome (ARDS). The objective of this study was to was more than the expected number if the samples were determine the similarity of the lung fluid samples recovered identical. There were nine proteins that were consistently by these methods using proteomic analysis. differentially expressed between the mini-BAL and s-Cath Methods Distal lung fluid samples were collected from samples. Of these nine proteins, five are abundantly found seven mechanically ventilated patients with ARDS using in neutrophils or airway epithelial cells, suggesting that the both s-Cath and mini-BAL in each patient and compared s-Cath may sample the bronchial airways to a greater extent using two-dimensional difference gel electrophoresis. -

Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities
REVIEW www.jasn.org Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities Chelsea C. Estrada,1 Alejandro Maldonado,1 and Sandeep K. Mallipattu1,2 1Division of Nephrology, Department of Medicine, Stony Brook University, Stony Brook, New York; and 2Renal Section, Northport Veterans Affairs Medical Center, Northport, New York ABSTRACT Inhibition of vascular endothelial growth factor A (VEGFA)/vascular endothelial with hypertension and proteinuria. Re- growth factor receptor 2 (VEGFR2) signaling is a common therapeutic strategy in ports describe histologic changes in the oncology, with new drugs continuously in development. In this review, we consider kidney primarily as glomerular endothe- the experimental and clinical evidence behind the diverse nephrotoxicities associ- lial injury with thrombotic microangiop- ated with the inhibition of this pathway. We also review the renal effects of VEGF athy (TMA).8 Nephrotic syndrome has inhibition’s mediation of key downstream signaling pathways, specifically MAPK/ also been observed,9 with the clinical ERK1/2, endothelial nitric oxide synthase, and mammalian target of rapamycin manifestations varying according to (mTOR). Direct VEGFA inhibition via antibody binding or VEGF trap (a soluble decoy mechanism and direct target of VEGF receptor) is associated with renal-specific thrombotic microangiopathy (TMA). Re- inhibition. ports also indicate that tyrosine kinase inhibition of the VEGF receptors is prefer- Current VEGF inhibitors can be clas- entially associated with glomerulopathies such as minimal change disease and FSGS. sifiedbytheirtargetofactioninthe Inhibition of the downstream pathway RAF/MAPK/ERK has largely been associated VEGFA-VEGFR2 pathway: drugs that with tubulointerstitial injury. Inhibition of mTOR is most commonly associated with bind to VEGFA, sequester VEGFA, in- albuminuria and podocyte injury, but has also been linked to renal-specificTMA.In hibit receptor tyrosine kinases (RTKs), all, we review the experimentally validated mechanisms by which VEGFA-VEGFR2 or inhibit downstream pathways. -
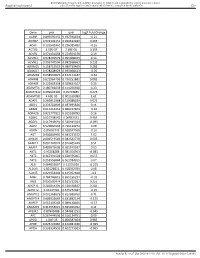
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Rage (Receptor for Advanced Glycation End Products) in Melanoma
RAGE (RECEPTOR FOR ADVANCED GLYCATION END PRODUCTS) IN MELANOMA PROGRESSION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Varsha Meghnani In Partial Fulfillment for the Degree of DOCTOR OF PHILOSOPHY Major Department: Pharmaceutical Sciences May 2014 Fargo, North Dakota North Dakota State University Graduate School Title RAGE (RECEPTOR FOR ADVANCED GLYCATION END PRODUCTS) IN MELANOMA PROGRESSION By VARSHA MEGHNANI The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: ESTELLE LECLERC Chair BIN GUO STEPHEN O’ROURKE JANE SCHUH Approved: 5/22/2014 JAGDISH SINGH Date Department Chair ABSTRACT The Receptor for Advanced Glycation End Products (RAGE) and its ligands are expressed in multiple cancer types and are implicated in cancer progression. Our lab has previously reported higher transcript levels of RAGE and S100B in advanced staged melanoma patients. The contribution of RAGE in the pathophysiology of melanoma has not been well studied. Based on previous reports, we hypothesized that RAGE, when over-expressed in melanoma cells, promotes melanoma progression. To study the pathogenic role of RAGE in melanoma, a primary melanoma cell line, WM115, was selected and stably transfected with full length RAGE (FL_RAGE) to generate a model cell line over-expressing RAGE (WM115_RAGE). WM266, a sister cell line of WM115, originated from a metastatic tumor of the same patient was used as a metastatic control cell line in the study. After assessing the expression levels of RAGE in the transfected cells, the influence of RAGE on their cellular properties was examined.