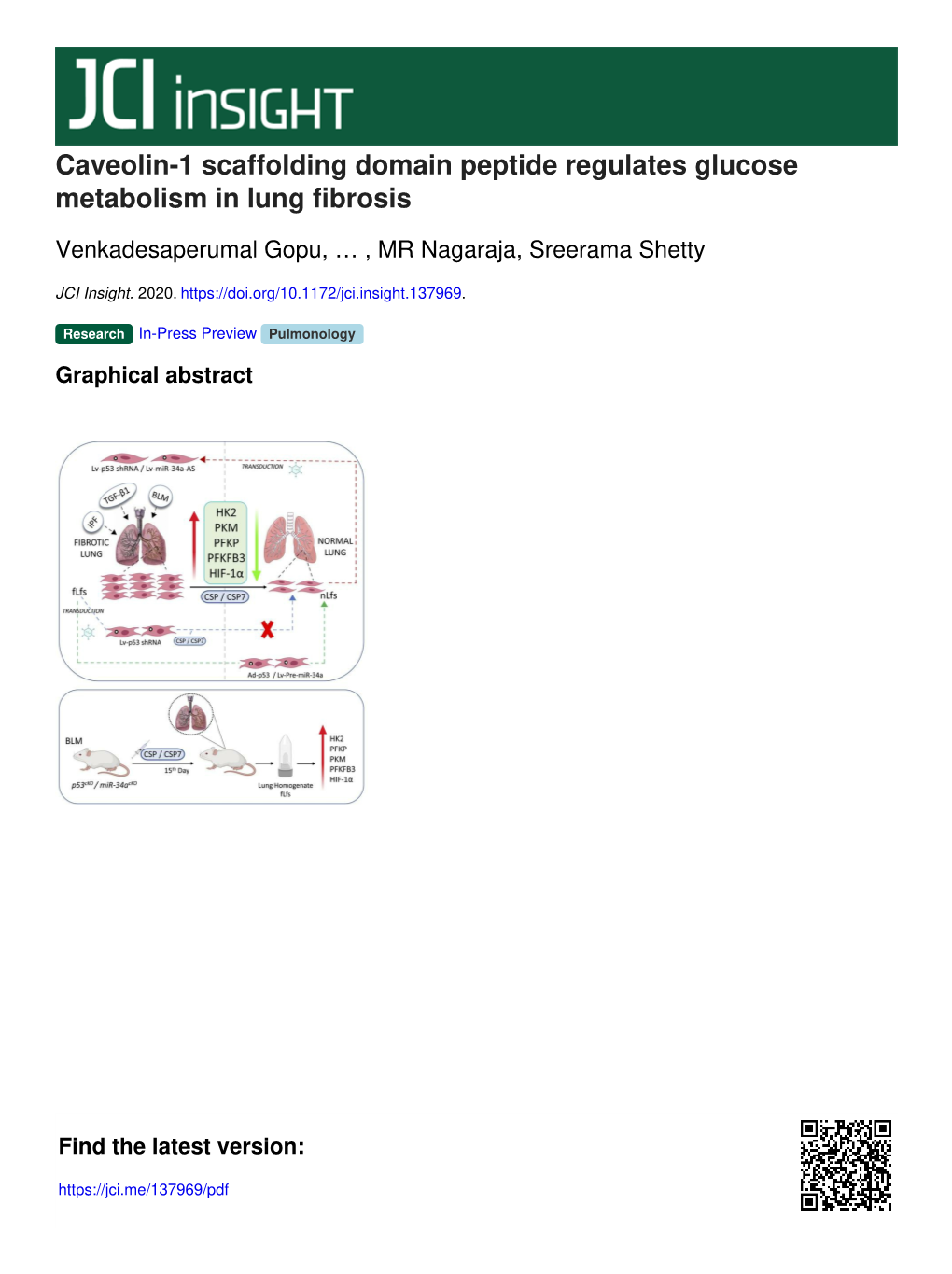
Caveolin-1 Scaffolding Domain Peptide Regulates Glucose Metabolism in Lung Fibrosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small-Molecule Inhibition of 6-Phosphofructo-2-Kinase Activity Suppresses Glycolytic Flux and Tumor Growth
110 Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth Brian Clem,1,3 Sucheta Telang,1,3 Amy Clem,1,3 reduces the intracellular concentration of Fru-2,6-BP, Abdullah Yalcin,1,2,3 Jason Meier,2 glucose uptake, and growth of established tumors in vivo. Alan Simmons,1,3 Mary Ann Rasku,1,3 Taken together, these data support the clinical development Sengodagounder Arumugam,1,3 of 3PO and other PFKFB3 inhibitors as chemotherapeutic William L. Dean,2,3 John Eaton,1,3 Andrew Lane,1,3 agents. [Mol Cancer Ther 2008;7(1):110–20] John O. Trent,1,2,3 and Jason Chesney1,2,3 Departments of 1Medicine and 2Biochemistry and Molecular Introduction Biology and 3Molecular Targets Group, James Graham Brown Neoplastic transformation causes a marked increase in Cancer Center, University of Louisville, Louisville, Kentucky glucose uptake and catabolic conversion to lactate, which forms the basis for the most specific cancer diagnostic 18 Abstract examination—positron emission tomography of 2- F- fluoro-2-deoxyglucose (18F-2-DG) uptake (1). The protein 6-Phosphofructo-1-kinase, a rate-limiting enzyme of products of several oncogenes directly increase glycolytic glycolysis, is activated in neoplastic cells by fructose-2,6- flux even under normoxic conditions, a phenomenon bisphosphate (Fru-2,6-BP), a product of four 6-phospho- originally termed the Warburg effect (2, 3). For example, fructo-2-kinase/fructose-2,6-bisphosphatase isozymes c-myc is a transcription factor that promotes the expression (PFKFB1-4). The inducible PFKFB3 isozyme is constitu- of glycolytic enzyme mRNAs, and its expression is increased tively expressed by neoplastic cells and required for the in several human cancers regardless of the oxygen pressure high glycolytic rate and anchorage-independent growth of (4, 5). -

Inhibition of BUB3 Shunts Glucose to Glycolytic Pathway by Inducing PFKFB3 Accumulation
Inhibition of BUB3 shunts glucose to glycolytic pathway by inducing PFKFB3 accumulation Jiajin Li ( [email protected] ) Shanghai Jiao Tong University School of Medicine Aliated Renji Hospital https://orcid.org/0000- 0003-1300-6025 Ruixue Zhang Shanghai Jiao Tong University School of Medicine Aliated Renji Hospital Gang Huang Shanghai Jiao Tong University School of Medicine Aliated Renji Hospital Jianjun Liu Shanghai Jiao Tong University School of Medicine Aliated Renji Hospital Research article Keywords: Glucose metabolism, Pentose phosphate pathway, Glycolysis, Cancer, BUB3, PFKFB3 Posted Date: October 25th, 2019 DOI: https://doi.org/10.21203/rs.2.16450/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/15 Abstract Purpose: Metabolic reprogramming as a hallmark of cancer has countless connections with other biological behavior of tumor such as rapid mitosis. Mitotic checkpoint protein BUB3 as a key protein involved in the regulation of mitosis is modulated by PKM2, an important glycolytic enzyme. However the role of BUB3 in glucose metabolism remains unknown. Methods: We analyzed the TCGA data to evaluate BUB3 expression in certain tumors. The uptake of glucose and CO2 incorporation was tested by isotopic tracer methods. The lactate, NADPH, NADP and metabolic enzyme activities were tested by assay kits accordingly. Results: We show here that BUB3 is over expressed in cervical cancer and hepatocellular carcinoma. Interference of BUB3 increase the uptake of glucose and shunts the metabolic ux from pentose phosphate pathway to glycolytic pathway. The glycolysis metabolites lactate is increased by BUB3 interference whereas NADPH/NADP ratio is reduced. -

Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: an Update
International Journal of Molecular Sciences Review Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update Philippe Icard 1,2,3,*, Antoine Coquerel 1,4, Zherui Wu 5 , Joseph Gligorov 6, David Fuks 7, Ludovic Fournel 3,8, Hubert Lincet 9,10 and Luca Simula 11 1 Medical School, Université Caen Normandie, CHU de Caen, 14000 Caen, France; [email protected] 2 UNICAEN, INSERM U1086 Interdisciplinary Research Unit for Cancer Prevention and Treatment, Normandie Université, 14000 Caen, France 3 Service de Chirurgie Thoracique, Hôpital Cochin, Hôpitaux Universitaires Paris Centre, APHP, Paris-Descartes University, 75014 Paris, France; [email protected] 4 INSERM U1075, COMETE Mobilités: Attention, Orientation, Chronobiologie, Université Caen, 14000 Caen, France 5 School of Medicine, Shenzhen University, Shenzhen 518000, China; [email protected] 6 Oncology Department, Tenon Hospital, Pierre et Marie Curie University, 75020 Paris, France; [email protected] 7 Service de Chirurgie Digestive et Hépato-Biliaire, Hôpital Cochin, Hôpitaux Universitaires Paris Centre, APHP, Paris-Descartes University, 75014 Paris, France; [email protected] 8 Descartes Faculty of Medicine, University of Paris, Paris Center, 75006 Paris, France 9 INSERM U1052, CNRS UMR5286, Cancer Research Center of Lyon (CRCL), 69008 Lyon, France; [email protected] 10 ISPB, Faculté de Pharmacie, Université Lyon 1, 69373 Lyon, France 11 Department of Infection, Immunity and Inflammation, Institut Cochin, INSERM U1016, CNRS UMR8104, Citation: Icard, P.; Coquerel, A.; Wu, University of Paris, 75014 Paris, France; [email protected] Z.; Gligorov, J.; Fuks, D.; Fournel, L.; * Correspondence: [email protected] Lincet, H.; Simula, L. -

Metabolic Regulation of Calcium Pumps in Pancreatic Cancer: Role of Phosphofructokinase-Fructose- Bisphosphatase-3 (PFKFB3) D
Richardson et al. Cancer & Metabolism (2020) 8:2 https://doi.org/10.1186/s40170-020-0210-2 RESEARCH Open Access Metabolic regulation of calcium pumps in pancreatic cancer: role of phosphofructokinase-fructose- bisphosphatase-3 (PFKFB3) D. A. Richardson1, P. Sritangos1, A. D. James2, A. Sultan1 and J. I. E. Bruce1* Abstract Background: High glycolytic rate is a hallmark of cancer (Warburg effect). Glycolytic ATP is required for fuelling plasma membrane calcium ATPases (PMCAs), responsible for extrusion of cytosolic calcium, in pancreatic ductal adenocarcinoma (PDAC). Phosphofructokinase-fructose-bisphosphatase-3 (PFKFB3) is a glycolytic driver that activates key rate-limiting enzyme Phosphofructokinase-1; we investigated whether PFKFB3 is required for PMCA function in PDAC cells. Methods: PDAC cell-lines, MIA PaCa-2, BxPC-3, PANC1 and non-cancerous human pancreatic stellate cells (HPSCs) were used. Cell growth, death and metabolism were assessed using sulforhodamine-B/tetrazolium-based assays, poly-ADP- ribose-polymerase (PARP1) cleavage and seahorse XF analysis, respectively. ATP was measured using a luciferase-based assay, membrane proteins were isolated using a kit and intracellular calcium concentration and PMCA activity were measured using Fura-2 fluorescence imaging. Results: PFKFB3 was highly expressed in PDAC cells but not HPSCs. In MIA PaCa-2, a pool of PFKFB3 was identified at the plasma membrane. PFKFB3 inhibitor, PFK15, caused reduced cell growth and PMCA activity, leading to calcium overload and apoptosis in PDAC cells. PFK15 reduced glycolysis but had noeffectonsteady-stateATPconcentrationinMIAPaCa-2. Conclusions: PFKFB3 is important for maintaining PMCA function in PDAC, independently of cytosolic ATP levels and may be involved in providing a localised ATP supply at the plasma membrane. -

Mir-206 Regulates a Metabolic Switch in Nasopharyngeal Carcinoma by Suppressing HK2 Expression
miR-206 regulates a metabolic switch in nasopharyngeal carcinoma by suppressing HK2 expression Chunying Luo Guangxi medical university Min Liu Shanghai 6th Peoples Hospital Aliated to Shanghai Jiaotong University School of Medicine Jianwei Zhang Shanghai 6th Peoples Hospital Aliated to Shanghai Jiaotong University School of Medicine Guoqiang Su Guangxi medical University Zhonghua Wei ( [email protected] ) Shanghai 6th Peoples Hospital Aliated to Shanghai Jiaotong University School of Medicine https://orcid.org/0000-0003-0011-2347 Research article Keywords: miR-206, Glycolysis, HK2, Nasopharyngeal Carcinoma Posted Date: July 22nd, 2020 DOI: https://doi.org/10.21203/rs.3.rs-43672/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/13 Abstract Background: Many studies have shown that microRNAs play key functions in nasopharyngeal carcinoma proliferation, invasion and metastasis. However, whether the dysregulated level of miRNAs contributes to the metabolic shift in nasopharyngeal carcinoma is not completely understood. Objectives: This study was conducted to explore the expression and function of miR-206 in nasopharyngeal carcinoma. Methods: miR-206 expression level was examined by real-time PCR. miR-206 inhibitor, mimics, and scrambled control were transiently transfected into nasopharyngeal carcinoma cells and their effects on colony formation, glucose uptake, and lactate secretion were observed in vitro. Moreover, the relationship between the levels of miR-206 and HK2 was examined by luciferase reporter and assay western blot. Results: In our study, we reported downregulation of miR-206 expression leads to metabolic change in nasopharyngeal carcinoma cells. miR-206 controls this function by enhancing HK2 expression. -

Compression-Induced Expression of Glycolysis Genes in Cafs Correlates with EMT and Angiogenesis Gene Expression in Breast Cancer
ARTICLE https://doi.org/10.1038/s42003-019-0553-9 OPEN Compression-induced expression of glycolysis genes in CAFs correlates with EMT and angiogenesis gene expression in breast cancer Baek Gil Kim 1,2, Jin Sol Sung2, Yeonsue Jang1, Yoon Jin Cha1, Suki Kang1,3, Hyun Ho Han2, Joo Hyun Lee2 & 1234567890():,; Nam Hoon Cho1,2,3,4 Tumor growth increases compressive stress within a tissue, which is associated with solid tumor progression. However, very little is known about how compressive stress contributes to tumor progression. Here, we show that compressive stress induces glycolysis in human breast cancer associated fibroblast (CAF) cells and thereby contributes to the expression of epithelial to mesenchymal (EMT)- and angiogenesis-related genes in breast cancer cells. Lactate production was increased in compressed CAF cells, in a manner dependent on the expression of metabolic genes ENO2, HK2, and PFKFB3. Conditioned medium from com- pressed CAFs promoted the proliferation of breast cancer cells and the expression of EMT and/or angiogenesis-related genes. In patient tissues with high compressive stress, the expression of compression-induced metabolic genes was significantly and positively corre- lated with EMT and/or angiogenesis-related gene expression and metastasis size. These findings illustrate a mechanotransduction pathway involving stromal glycolysis that may be relevant also for other solid tumours. 1 Department of Pathology, Yonsei University College of Medicine, Seoul, South Korea. 2 Brain Korea 21 Plus Project for Medical Science, Yonsei University College of Medicine, Seoul, South Korea. 3 Severance Biomedical Science Institute (SBSI), Yonsei University College of Medicine, Seoul, South Korea. 4 Global 5-5-10 System Biology, Yonsei University, Seoul, South Korea. -

The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis Via GPER1 S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2017/03/27/jpet.116.238212.DC1 1521-0103/361/3/398–407$25.00 https://doi.org/10.1124/jpet.116.238212 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 361:398–407, June 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1 s Annalisa Trenti, Serena Tedesco, Carlotta Boscaro, Nicola Ferri, Andrea Cignarella, Lucia Trevisi, and Chiara Bolego Department of Pharmaceutical and Pharmacological Sciences (A.T., S.T., Ca.B., N.F., L.T., Ch.B) and Department of Medicine (A.C.), University of Padova, Padova, Italy Received October 6, 2016; accepted March 22, 2017 Downloaded from ABSTRACT The endogenous estrogen 17b-estradiol (E2) is a key factor in vein endothelial cells (HUVECs); in addition, E2 treatment promoting endothelial healing and angiogenesis. Recently, upregulated PFKFB3 expression in a time- and concentration- proangiogenic signals including vascular endothelial growth dependent manner. Such an effect peaked at 3 hours and was factor and others have been shown to converge in endothelial also induced by G-1 and abolished by pretreatment with the cell metabolism. Because inhibition of the glycolytic enzyme GPER1 antagonist G-15 or GPER1 siRNA, consistent with activator phosphofructokinase-2/fructose-2,6-bisphosphatase engagement of membrane ER. Experiments with the PFKFB3 jpet.aspetjournals.org 3 (PFKFB3) reduces pathologic angiogenesis and estrogen inhibitor 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one showed receptor (ER) signaling stimulates glucose uptake and glycolysis that PFKFB3 activity was required for estrogen-mediated by inducing PFKFB3 in breast cancer, we hypothesized that E2 HUVEC migration via GPER1. -

How Do Glycolytic Enzymes Favour Cancer Cell Proliferation by Nonmetabolic Functions?
Oncogene (2015) 34, 3751–3759 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? H Lincet1,2,3 and P Icard1,4 Cancer cells enhance their glycolysis, producing lactate, even in the presence of oxygen. Glycolysis is a series of ten metabolic reactions catalysed by enzymes whose expression is most often increased in tumour cells. HKII and phosphoglucose isomerase (PGI) have mainly an antiapoptotic effect; PGI and glyceraldehyde-3-phosphate dehydrogenase activate survival pathways (Akt and so on); phosphofructokinase 1 and triose phosphate isomerase participate in cell cycle activation; aldolase promotes epithelial mesenchymal transition; PKM2 enhances various nuclear effects such as transcription, stabilisation and so on. This review outlines the multiple non-glycolytic roles of glycolytic enzymes, which are essential for promoting cancer cells' survival, proliferation, chemoresistance and dissemination. Oncogene (2015) 34, 3751–3759; doi:10.1038/onc.2014.320; published online 29 September 2014 INTRODUCTION implications in many other functions, such as apoptosis, In normal tissue, the vast majority of nonproliferating differen- detoxification, cell cycle control, signalling pathways and so on. tiated cells use oxidative phosphorylation (OXPHOS) for ATP production. These cells metabolise glucose to pyruvate through Hexokinases glycolysis, then oxidise this pyruvate through the tricarboxylic acid In the cytosol, glucose (or fructose) is phosphorylated by cycle, generating ATP through ATP synthase, the rate of the hexokinases (HK) (glucose kinase or fructose kinase) to glucose- production being coupled with proton transport and on oxygen 6-phosphate (G6P). HK catalye the first irreversible reaction of respiration.1 In contrast, rapidly proliferating tumour cells con- glycolysis. -

Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential As Therapeutic Targets
cancers Review Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets Krzysztof Kotowski 1 , Jakub Rosik 2 , Filip Machaj 2, Stanisław Supplitt 3 , Daniel Wiczew 4,5 , Karolina Jabło ´nska 1 , Emilia Wiechec 6,7, Saeid Ghavami 8,9,* and Piotr Dzi˛egiel 1,10,* 1 Department of Histology and Embryology, Wroclaw Medical University, 50-368 Wroclaw, Poland; [email protected] (K.K.); [email protected] (K.J.) 2 Department of Pathology, Pomeranian Medical University, 71-252 Szczecin, Poland; [email protected] (J.R.); [email protected] (F.M.) 3 Department of Genetics, Wroclaw Medical University, 50-368 Wroclaw, Poland; [email protected] 4 Department of Biochemical Engineering, Wroclaw University of Science and Technology, 50-370 Wroclaw, Poland; [email protected] 5 Laboratoire de physique et chimie théoriques, Université de Lorraine, F-54000 Nancy, France 6 Department of Biomedical and Clinical Sciences (BKV), Division of Cell Biology, Linköping University, Region Östergötland, 581 85 Linköping, Sweden; [email protected] 7 Department of Otorhinolaryngology in Linköping, Anesthetics, Operations and Specialty Surgery Center, Region Östergötland, 581 85 Linköping, Sweden 8 Department of Human Anatomy and Cell Science, Rady Faculty of Health Sciences, Max Rady College of Medicine, University of Manitoba, Winnipeg, MB R3E 0J9, Canada Citation: Kotowski, K.; Rosik, J.; 9 Research Institute in Oncology and Hematology, Cancer Care Manitoba, University of Manitoba, Machaj, F.; Supplitt, S.; Wiczew, D.; Winnipeg, MB R3E 0V9, Canada Jabło´nska,K.; Wiechec, E.; Ghavami, 10 Department of Physiotherapy, Wroclaw University School of Physical Education, 51-612 Wroclaw, Poland S.; Dzi˛egiel,P. -

Mir-302 Regulates Glycolysis to Control Cell-Cycle During Neural Tube Closure
International Journal of Molecular Sciences Article MiR-302 Regulates Glycolysis to Control Cell-Cycle during Neural Tube Closure Rachel A. Keuls 1,2 , Karin Kojima 2, Brittney Lozzi 3, John W. Steele 4,5 , Qiuying Chen 6, Steven S. Gross 6, Richard H. Finnell 4,5 and Ronald J. Parchem 1,2,4,* 1 Development, Disease Models & Therapeutics Graduate Program, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA; [email protected] 2 Center for Cell and Gene Therapy, Stem Cells and Regenerative Medicine Center, One Baylor Plaza, Houston, TX 77030, USA; [email protected] 3 Genetics and Genomics Graduate Program, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA; [email protected] 4 Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX 77030, USA; [email protected] (J.W.S.); [email protected] (R.H.F.) 5 Center for Precision Environmental Health, Department of Molecular and Cellular Biology and Medicine, Baylor College of Medicine, Houston, TX 77030, USA 6 Department of Pharmacology, Weill Cornell Medicine, New York, NY 10065, USA; [email protected] (Q.C.); [email protected] (S.S.G.) * Correspondence: [email protected] Received: 30 August 2020; Accepted: 6 October 2020; Published: 13 October 2020 Abstract: Neural tube closure is a critical early step in central nervous system development that requires precise control of metabolism to ensure proper cellular proliferation and differentiation. Dysregulation of glucose metabolism during pregnancy has been associated with neural tube closure defects (NTDs) in humans suggesting that the developing neuroepithelium is particularly sensitive to metabolic changes. -

GAPDH Enhances the Aggressiveness and the Vascularization of Non-Hodgkin’S B Lymphomas Via NF-Κb-Dependent Induction of HIF-1Α
Leukemia (2015) 29, 1163–1176 © 2015 Macmillan Publishers Limited All rights reserved 0887-6924/15 www.nature.com/leu ORIGINAL ARTICLE GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-κB-dependent induction of HIF-1α J Chiche1,2, S Pommier1,2,3, M Beneteau1,2, L Mondragón1,2, O Meynet1,2, B Zunino1,2, A Mouchotte1,2, E Verhoeyen1,2, M Guyot2,4, G Pagès2,4, N Mounier5, V Imbert2,6, P Colosetti2,7, D Goncalvès2,7, S Marchetti2,7, J Brière8, M Carles1,2,3, C Thieblemont8 and J-E Ricci1,2,3 Deregulated expression of glycolytic enzymes contributes not only to the increased energy demands of transformed cells but also has non-glycolytic roles in tumors. However, the contribution of these non-glycolytic functions in tumor progression remains poorly defined. Here, we show that elevated expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), but not of other glycolytic enzymes tested, increased aggressiveness and vascularization of non-Hodgkin’s lymphoma. Elevated GAPDH expression was found to promote nuclear factor-κB (NF-κB) activation via binding to tumor necrosis factor receptor-associated factor-2 (TRAF2), enhancing the transcription and the activity of hypoxia-inducing factor-1α (HIF-1α). Consistent with this, inactive mutants of GAPDH failed to bind TRAF2, enhance HIF-1 activity or promote lymphomagenesis. Furthermore, elevated expression of gapdh mRNA in biopsies from diffuse large B-cell non-Hodgkin’s lymphoma patients correlated with high levels of hif-1α, vegf-a, nfkbia mRNA and CD31 staining. Collectively, these data indicate that deregulated GAPDH expression promotes NF-κB-dependent induction of HIF-1α and has a key role in lymphoma vascularization and aggressiveness. -

PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair
cancers Article PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair Anna Huguet Ninou 1,2, Jemina Lehto 1,2 , Dimitrios Chioureas 1, Hannah Stigsdotter 1, Korbinian Schelzig 1, Emma Åkerlund 1, Greta Gudoityte 1 , Ulrika Joneborg 3, Joseph Carlson 4,5, Jos Jonkers 6 , Brinton Seashore-Ludlow 1 and Nina Marie Susanne Gustafsson 1,* 1 Science for Life Laboratory, Department of Oncology and Pathology, Karolinska Institutet, 171 21 Stockholm, Sweden; [email protected] (A.H.N.); [email protected] (J.L.); [email protected] (D.C.); [email protected] (H.S.); [email protected] (K.S.); [email protected] (E.Å.); [email protected] (G.G.); [email protected] (B.S.-L.) 2 Kancera AB, Karolinska Science Park, 171 48 Solna, Sweden 3 Department of Women’s and Children’s Health, Karolinska Institutet, 171 21 Stockholm, Sweden; [email protected] 4 Department of Oncology and Pathology, Karolinska Institutet, 171 76 Stockholm, Sweden; [email protected] 5 Department of Pathology and Laboratory Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90089, USA 6 Oncode Institute and Division of Molecular Pathology, The Netherlands Cancer Institute, 1066CX Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected] Simple Summary: DNA-damaging chemotherapeutics, such as platinum drugs, are cornerstones Citation: Ninou, A.H.; Lehto, J.; Chioureas, D.; Stigsdotter, H.; in cancer treatment. The efficacy of such treatment is intimately linked to the DNA repair capacity Schelzig, K.; Åkerlund, E.; Gudoityte, of the cancer cells, as DNA damage above a tolerable threshold culminates in cell death.