BGGN-213: FOUNDATIONS of BIOINFORMATICS (Lecture 17) Biological Network Analysis (Part 1)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Simplified Computational Model for Generating Bio- Logical Networks
Simplified Computational Model for Generating Bio- logical Networks† Matthew H J Bailey,∗ David Ormrod Morley,∗ and Mark Wilson∗ A method to generate and simulate biological networks is discussed. An expanded Wooten- Winer-Weaire bond switching methods is proposed which allows for a distribution of node degrees in the network while conserving the mean average node degree. The networks are characterised in terms of their polygon structure and assortativities (a measure of local ordering). A wide range of experimental images are analysed and the underlying networks quantified in an analogous manner. Limitations in obtaining the network structure are discussed. A “network landscape” of the experimentally observed and simulated networks is constructed from the underlying metrics. The enhanced bond switching algorithm is able to generate networks spanning the full range of experimental observations. Two dimensional random networks are observed in a range of 17 contexts across considerably different length scales in nature: to be limited to avoid damaging the delicate networks . How- from nanometres, in the form of amorphous graphene; to me- ever, even when a high-quality image is obtained, there is further tres, in the form of the Giant’s causeway; to tens of kilometres, in difficulty in analysis. For example, each edge in a network is a the form of geopolitical borders 1–3. A framework for describing complex molecule made up of tens of thousands of atoms which these continuous random networks for chemical systems was first may not lie strictly in a single plane. In addition, the most inter- introduced by Zachariasen to describe silica-like glasses, and has esting dynamic behaviour often occurs over very long timescales proved to be extremely versatile in the years since 4, being used to – potentially decades. -

Biological Network Approaches and Applications in Rare Disease Studies
G C A T T A C G G C A T genes Review Biological Network Approaches and Applications in Rare Disease Studies Peng Zhang 1,* and Yuval Itan 2,3 1 St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065, USA 2 The Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; [email protected] 3 Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA * Correspondence: [email protected]; Tel.: +1-646-830-6622 Received: 3 September 2019; Accepted: 10 October 2019; Published: 12 October 2019 Abstract: Network biology has the capability to integrate, represent, interpret, and model complex biological systems by collectively accommodating biological omics data, biological interactions and associations, graph theory, statistical measures, and visualizations. Biological networks have recently been shown to be very useful for studies that decipher biological mechanisms and disease etiologies and for studies that predict therapeutic responses, at both the molecular and system levels. In this review, we briefly summarize the general framework of biological network studies, including data resources, network construction methods, statistical measures, network topological properties, and visualization tools. We also introduce several recent biological network applications and methods for the studies of rare diseases. Keywords: biological network; bioinformatics; database; software; application; rare diseases 1. Introduction Network biology provides insights into complex biological systems and can reveal informative patterns within these systems through the integration of biological omics data (e.g., genome, transcriptome, proteome, and metabolome) and biological interactome data (e.g., protein-protein interactions and gene-gene associations). -

Denoising Large-Scale Biological Data Using Network Filters
Kavran and Clauset BMC Bioinformatics (2021) 22:157 https://doi.org/10.1186/s12859‑021‑04075‑x RESEARCH ARTICLE Open Access Denoising large‑scale biological data using network flters Andrew J. Kavran1,2 and Aaron Clauset2,3,4* *Correspondence: [email protected] Abstract 2 BioFrontiers Institute, Background: Large-scale biological data sets are often contaminated by noise, which University of Colorado, Boulder, CO, USA can impede accurate inferences about underlying processes. Such measurement Full list of author information noise can arise from endogenous biological factors like cell cycle and life history vari- is available at the end of the ation, and from exogenous technical factors like sample preparation and instrument article variation. Results: We describe a general method for automatically reducing noise in large- scale biological data sets. This method uses an interaction network to identify groups of correlated or anti-correlated measurements that can be combined or “fltered” to better recover an underlying biological signal. Similar to the process of denoising an image, a single network flter may be applied to an entire system, or the system may be frst decomposed into distinct modules and a diferent flter applied to each. Applied to synthetic data with known network structure and signal, network flters accurately reduce noise across a wide range of noise levels and structures. Applied to a machine learning task of predicting changes in human protein expression in healthy and cancer- ous tissues, network fltering prior to training increases accuracy up to 43% compared to using unfltered data. Conclusions: Network flters are a general way to denoise biological data and can account for both correlation and anti-correlation between diferent measurements. -

Introduction to Graph Theory
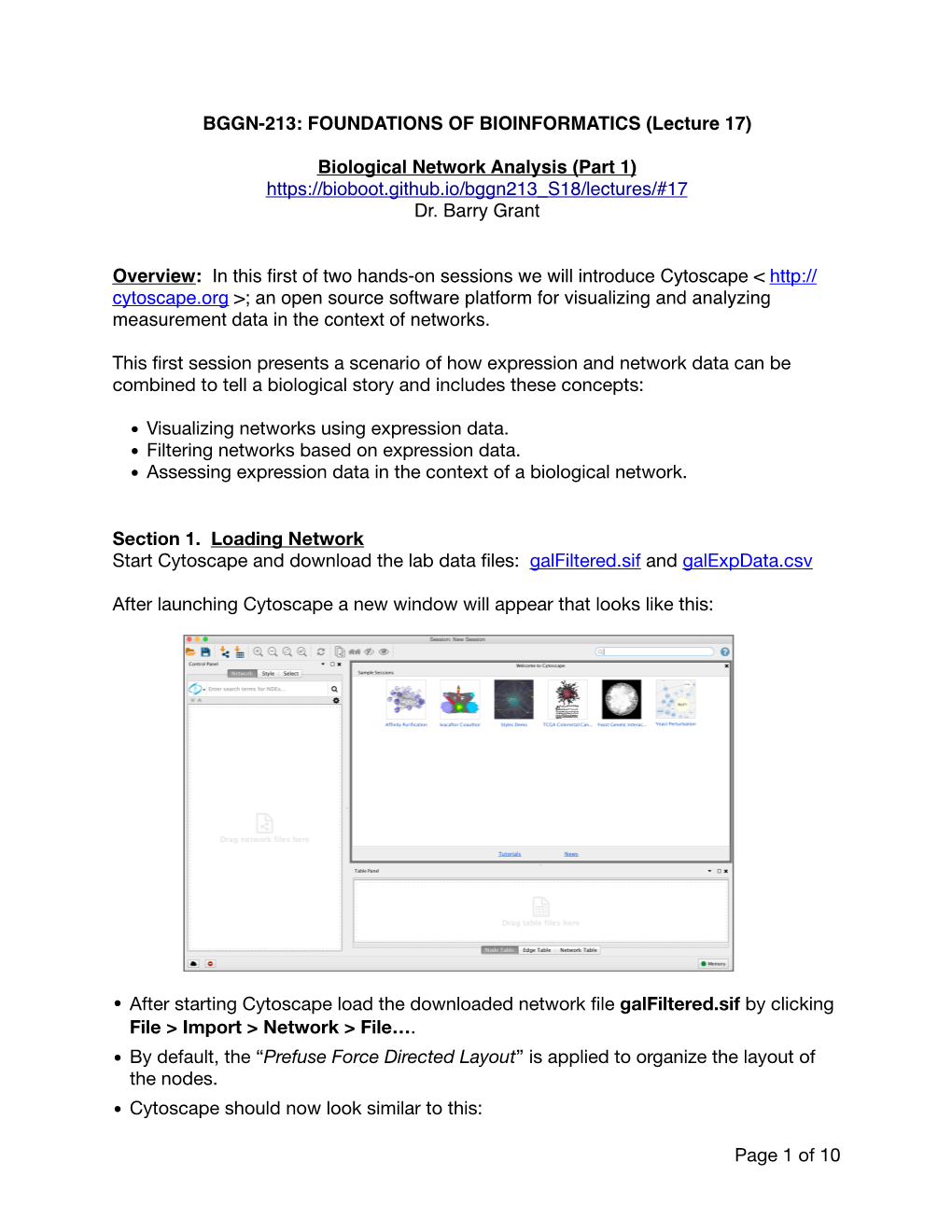
Introduction to Graph Theory Proteomes Interactomes and Biological Networks Department of Pharmacy and Emidio Capriotti Biotechnology (FaBiT) http://biofold.org/ University of Bologna Historical Perspective With the Seven Bridges of Königsberg problem, Euler in 1737 laid the foundations of the graph theory. Simon Kneebone – simonkneebone.com • Find path (Eulerian Path) that traverses all the Pregel’s bridges. • Find walk (Eulerian Circuit) that traverses all the Pregel’s bridges and has the same starting and ending point. Solution Describe the problem as a graph where the nodes represent the 4 locations and the edges correspond to the bridges Northern Bank Island 1 Island 2 Southern Bank Eulerian path exists only if zero or 2 nodes are connected by an odd number of bridges. Eulerian circuit exists only if zero nodes are connected by an odd number of bridges. Graph Definition A graph is a pair G=(V,E) consisting of two sets: • V is a set of elements called Nodes or Vertices. • E is a set of pairs (vi,vj) where vi∊V and vj∊V. The pairs E are links between two nodes and are called Edges A V = {A; B; C; D} B C E = {(A,B); (A,C); (B,C); (B,D); (C,D)} D Undirected Graph Undirected graph is a network where the relationship between nodes are symmetric. D V = {Group of People} A B C E E = {Pairs of Friends} F Directed Graph Directed graph is a network where the relationship between nodes are asymmetric. In this case the edges are directed lines. B C V = {Group of Animals} A D E E = {Pray/Predator Relationships} F G Signed Directed Graph Signed Directed graph is a network where the relationship between nodes are asymmetric and have positive or negative associated signs RecKinase RecCAMP_act RecCAMP_de Galpha_bnd Galpha_act_bnd + − C E − V = {Group of Genes} A B + E = {Activation/Inhibition Relationships} D Graph and Networks Graphs can be used to represent any observed network. -

Visual Analytics for Multimodal Social Network Analysis: a Design Study with Social Scientists
Visual Analytics for Multimodal Social Network Analysis: A Design Study with Social Scientists Sohaib Ghani, Student member, IEEE, Bum chul Kwon, Student member, IEEE, Seungyoon Lee, Ji Soo Yi, Member, IEEE, and Niklas Elmqvist, Senior member, IEEE Fig. 1. Four design sketches for visualizing multimodal social networks, evolved through discussion between social network experts and visual analytics experts. Early designs (top two) focus on splitting node-link diagrams into separate spaces, whereas the latter (bottom left) use vertical bands while maintaining compatibility with node-link diagrams (bottom right). Abstract—Social network analysis (SNA) is becoming increasingly concerned not only with actors and their relations, but also with distinguishing between different types of such entities. For example, social scientists may want to investigate asymmetric relations in organizations with strict chains of command, or incorporate non-actors such as conferences and projects when analyzing co- authorship patterns. Multimodal social networks are those where actors and relations belong to different types, or modes, and multimodal social network analysis (mSNA) is accordingly SNA for such networks. In this paper, we present a design study that we conducted with several social scientist collaborators on how to support mSNA using visual analytics tools. Based on an open- ended, formative design process, we devised a visual representation called parallel node-link bands (PNLBs) that splits modes into separate bands and renders connections between adjacent ones, similar to the list view in Jigsaw. We then used the tool in a qualitative evaluation involving five social scientists whose feedback informed a second design phase that incorporated additional network metrics. -

An Integrative Approach to Modeling Biological Networks 1 Introduction
Journal of Integrative Bioinformatics, 7(3):120, 2010 http://journal.imbio.de An integrative approach to modeling biological networks Vesna Memisevˇ ic´ 1, Tijana Milenkovic´ 1, and Natasaˇ Prˇzulj2,∗ 1Department of Computer Science, University of California, Irvine, CA 92697-3435, USA 2Department of Computing, Imperial College London, London, SW7 2AZ, UK ∗Corresponding author (e-mail: [email protected]) Summary Networks are used to model real-world phenomena in various domains, including systems biology. Since proteins carry out biological processes by interacting with other proteins, it is expected that cellular functions are reflected in the structure of protein-protein inter- action (PPI) networks. Similarly, the topology of residue interaction graphs (RIGs) that model proteins’ 3-dimensional structure might provide insights into protein folding, sta- bility, and function. An important step towards understanding these networks is finding an adequate network model, since models can be exploited algorithmically as well as used for predicting missing data. Evaluating the fit of a model network to the data is a formidable challenge, since network comparisons are computationally infeasible and thus have to rely on heuristics, or “network properties.” We show that it is difficult to assess the reliability of the fit of a model using any network property alone. Thus, we present an integrative approach that feeds a variety of network properties into five machine learning classifiers to predict the best-fitting network model for PPI networks and RIGs. We confirm that ge- ometric random graphs (GEO) are the best-fitting model for RIGs. Since GEO networks model spatial relationships between objects and are thus expected to replicate well the un- derlying structure of spatially packed residues in a protein, the good fit of GEO to RIGs validates our approach. -

Communities and Homology in Protein-Protein Interactions
Communities and Homology in Protein-protein Interactions Anna Lewis Balliol College University of Oxford A thesis submitted for the degree of Doctor of Philosophy Michaelmas 2011 2 3 This thesis is dedicated to the inspirational Jen Weterings. Wishing her well in her ongoing recovery. Acknowledgements With thanks to Charlotte, Nick and Mason, for being the best supervisors a girl could ask for. With thanks to all those who have helped me with the work in this thesis. In particular Sumeet Agarwal, Rebecca Hamer, Gesine Reinert, James Wakefield, Simon Myers, Steve Kelly, Nicholas Lewis, members of the Oxford Protein Informatics Group and members of the Oxford/Imperial Systems and Signalling group. With thanks to all those who have made my DPhil years a wonderful experience, in particular my family, my Balliol family, and my DTC family. Abstract Knowledge of protein sequences has exploded, but knowledge of protein function is needed to make use of sequence information, and this lags behind. A protein’s function must be understood in context and part of this is the network of interactions between proteins. What are the relationships between protein function and the structure of the interaction network? In the first part of my thesis, I investigate the functional relevance of clusters, or communities, of proteins in the yeast protein interaction network. Communities are candidates for biological modules. The work I present is the first to systematically investigate this structure at multiple scales in such networks. I develop novel tests to assess whether communities are functionally homogeneous, and demonstrate that almost every protein is found in a functionally homogeneous community at some scale. -

The Structure and Function of Complex Networks∗
SIAM REVIEW c 2003 Society for Industrial and Applied Mathematics Vol. 45,No. 2,pp. 167–256 The Structure and Function of Complex Networks∗ M. E. J. Newman† Abstract. Inspired by empirical studies of networked systems such as the Internet, social networks, and biological networks, researchers have in recent years developed a variety of techniques and models to help us understand or predict the behavior of these systems. Here we review developments in this field, including such concepts as the small-world effect, degree distributions, clustering, network correlations, random graph models, models of network growth and preferential attachment, and dynamical processes taking place on networks. Key words. networks, graph theory, complex systems, computer networks, social networks, random graphs, percolation theory AMS subject classifications. 05C75, 05C90, 94C15 PII. S0036144503424804 Contents. 1 Introduction 168 1.1 Types of Networks ............................. 171 1.2 Other Resources .............................. 172 1.3 Outline of the Review ........................... 174 2 Networks in the Real World 174 2.1 Social Networks ............................... 174 2.2 Information Networks ........................... 176 2.3 Technological Networks .......................... 178 2.4 Biological Networks ............................ 179 3 Properties of Networks 180 3.1 The Small-World Effect .......................... 180 3.2 Transitivity or Clustering ......................... 183 3.3 Degree Distributions ............................ 185 3.3.1 Scale-Free Networks ........................ 186 3.3.2 Maximum Degree .......................... 188 3.4 Network Resilience ............................. 189 3.5 Mixing Patterns .............................. 190 ∗Received by the editors January 20, 2003; accepted for publication (in revised form) March 17, 2003; published electronically May 2, 2003. This work was supported in part by the U.S. National Science Foundation under grants DMS-0109086 and DMS-0234188 and by the James S. -

'Small-World-Ness': a Quantitative Method for Determining Canonical Network Equivalence
This is a repository copy of Network 'small-world-ness': a quantitative method for determining canonical network equivalence. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/10308/ Article: Humphries, M.D. and Gurney, K. (2008) Network 'small-world-ness': a quantitative method for determining canonical network equivalence. Plos One, 3 (4). Art No.e0002051. ISSN 1932-6203 https://doi.org/10.1371/journal.pone.0002051 Reuse Unless indicated otherwise, fulltext items are protected by copyright with all rights reserved. The copyright exception in section 29 of the Copyright, Designs and Patents Act 1988 allows the making of a single copy solely for the purpose of non-commercial research or private study within the limits of fair dealing. The publisher or other rights-holder may allow further reproduction and re-use of this version - refer to the White Rose Research Online record for this item. Where records identify the publisher as the copyright holder, users can verify any specific terms of use on the publisher’s website. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Network ‘Small-World-Ness’: A Quantitative Method for Determining Canonical Network Equivalence Mark D. Humphries*, Kevin Gurney Adaptive Behaviour Research Group, Department of Psychology, University of Sheffield, Sheffield, United Kingdom Abstract Background: Many technological, biological, social, and information networks fall into the broad class of ‘small-world’ networks: they have tightly interconnected clusters of nodes, and a shortest mean path length that is similar to a matched random graph (same number of nodes and edges). -

Coupling AI and Network Biology
Coupling AI and network biology Generate insights for disease understanding and target identification Alexandr Ivliev, Director of Bioinformatics Cheng Fang, Scientific Consultant 03.06.2020 Agenda 1. Introduction 2. Coupling AI and network biology 3. High-quality biological networks in microbiome for target ID 4. Key takeaways Introduction © 2020 Clarivate 3 Genomic revolution of early 2000’s From individual genes to understanding entire genome © 2020 Clarivate 4 Artificial intelligence: new ongoing revolution Revolutionizing industries Computer Natural language Reinforcement vision processing learning • Self-driving cars • Machine translation • Chess, go, computer games • Face recognition • Speech analysis • Robotics • And more • And more • And more © 2020 Clarivate 5 Artificial intelligence is a hot field First time “deep learning” appeared in Gartner Hype Cycle for Emerging Technologies in 2017 © 2020 Clarivate 6 Deep learning is a big field https://www.asimovinstitute.org/neural-network-zoo/ © 2020 Clarivate 7 Computer vision and image processing Esteva et al, Nat Med, 2019 © 2020 Clarivate 8 Text mining, e.g. electronic health records Esteva et al, Nat Med, 2019 © 2020 Clarivate 9 Applications in genomics Esteva et al, Nat Med, 2019 © 2020 Clarivate 10 Networks are how biology works Drug target • Disease mechanism understanding Disease genes • Target ID Network by Martin Grandjean © 2020 Clarivate 11 Can biological networks be coupled with deep neural networks to enable disease mechanism understanding and target ID? Target © 2020 Clarivate 12 What approaches are you taking to understand disease mechanisms and identify novel drug targets? a. Literature searches b. OMICs data analysis c. Small scale lab experiments d. Classical machine learning e. Deep learning f. -

Network Biology: Understanding the Cell’S Functional Organization
REVIEWS NETWORK BIOLOGY: UNDERSTANDING THE CELL’S FUNCTIONAL ORGANIZATION Albert-László Barabási* & Zoltán N. Oltvai‡ A key aim of postgenomic biomedical research is to systematically catalogue all molecules and their interactions within a living cell. There is a clear need to understand how these molecules and the interactions between them determine the function of this enormously complex machinery, both in isolation and when surrounded by other cells. Rapid advances in network biology indicate that cellular networks are governed by universal laws and offer a new conceptual framework that could potentially revolutionize our view of biology and disease pathologies in the twenty-first century. PROTEIN CHIPS Reductionism, which has dominated biological research programme to map out, understand and model in quan- Similar to cDNA microarrays, for over a century, has provided a wealth of knowledge tifiable terms the topological and dynamic properties of the this evolving technology about individual cellular components and their func- various networks that control the behaviour of the cell. involves arraying a genomic set tions. Despite its enormous success, it is increasingly Help along the way is provided by the rapidly develop- of proteins on a solid surface without denaturing them. The clear that a discrete biological function can only rarely ing theory of complex networks that, in the past few proteins are arrayed at a high be attributed to an individual molecule. Instead, most years, has made advances towards uncovering the orga- enough density for the biological characteristics arise from complex interac- nizing principles that govern the formation and evolution detection of activity, binding tions between the cell’s numerous constituents, such as of various complex technological and social networks9–12. -

Biological Network Analysis Has Become a Central Component of Computational and Systems Biology
Pacific Symposium on Biocomputing 15:120-122(2010) DYNAMICS OF BIOLOGICAL NETWORKS: SESSION INTRODUCTION TANYA Y. BERGER-WOLF1 Department of Computer Science, University of Illinois at Chicago Chicago IL 60607, USA TERESA M. PRZYTYCKA2 National Center of Biotechnology Information, NLM, NIH Bethesda MD 20814, USA MONA SINGH 3 Department of Computer Science, Lewis Sigler Institute for Integrative Genomics Princeton University, Princeton NJ 08544, USA DONNA K. SLONIM 4 Department of Computer Science, Tufts University Department of Pathology, Tufts University School of Medicine Medford, MA 02155 Biological network analysis has become a central component of computational and systems biology. Because such analysis provides a unifying language to describe relations within complex systems, it has played an increasingly important role in understanding physiological function. Significant efforts have focused on analyzing and inferring the topology and structure of cellular networks and on relating them to cellular function and organization. However, much of this work has taken a static view of cellular networks, despite the knowledge that biological networks can change with time, context, and conditions. We introduce this session on The Dynamics of Biological Networks to encourage and support the development of computational methods that elucidate the dynamic interactome. Biological networks encompass many types of variation. Temporal variation can be inferred via experimentally determined large-scale (static) cellular networks, along with other high- throughput experimental data sets that provide snapshots of biological systems at different times and conditions. These data are often integrated with static interaction data (e.g. protein-protein, domain-domain, or regulatory interactions), time- or environment-dependent expression data, protein localization data, or other contextual information.