Cancer Subtype Identification Using Somatic Mutation Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Suppression of Signal Transducer and Activator of Transcription 3 Activation by Butein Inhibits Growth of Human Hepatocellular Carcinoma in Vivo
Author Manuscript Published OnlineFirst on December 3, 2010; DOI: 10.1158/1078-0432.CCR-10-1123 AuthorPublished manuscripts OnlineFirst have been on peer December reviewed and 3, accepted 2010 as for 10.1158/1078-0432.CCR-10-1123 publication but have not yet been edited. Suppression of Signal Transducer and Activator of Transcription 3 Activation by Butein Inhibits Growth of Human Hepatocellular Carcinoma in vivo Peramaiyan Rajendran1, Tina H. Ong2, Luxi Chen1,3, Feng Li1, Muthu K Shanmugam1, Shireen Vali4, Taher Abbasi4, Shweta Kapoor4, Ashish Sharma4, Alan Prem Kumar1,3, Kam M. Hui2,5 Gautam Sethi1,5 1Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, 2Division of Cellular and Molecular Research, Humphrey Oei Institute of Cancer Research National Cancer Centre, Singapore 169610, 3Cancer Science Institute of Singapore, National University of Singapore, and 4Cellworks Group Inc., California 95070; 4Cellworks Research India Pvt. Ltd, Bangalore 560066, India. Running title: Butein inhibits STAT3 signaling in vitro and in vivo in HCC. 5To whom correspondence should be addressed: 1. Dr. Gautam Sethi, Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Phone: +65-65163267; Fax: +65- 68737690; Email: [email protected] Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Copyright © 2010 American Association for Cancer Research Downloaded from clincancerres.aacrjournals.org on September 27, 2021. © 2010 American Association for Cancer Research. Author Manuscript Published OnlineFirst on December 3, 2010; DOI: 10.1158/1078-0432.CCR-10-1123 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Biological Pathways and in Vivo Antitumor Activity Induced by Atiprimod in Myeloma
Leukemia (2007) 21, 2519–2526 & 2007 Nature Publishing Group All rights reserved 0887-6924/07 $30.00 www.nature.com/leu ORIGINAL ARTICLE Biological pathways and in vivo antitumor activity induced by Atiprimod in myeloma P Neri1,2,3, P Tassone1,2,3, M Shammas1, H Yasui2, E Schipani4, RB Batchu1, S Blotta1,2,3, R Prabhala1, L Catley2, M Hamasaki2, T Hideshima2, D Chauhan2, GS Jacob5, D Picker5, S Venuta3, KC Anderson2 and NC Munshi1,2 1Jerome Lipper Multiple Myeloma Center, Department of Adult Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA; 2Boston VA Healthcare System, Department of Medicine, Harvard Medical School, MA, USA; 3Department of Experimental and Clinical Medicine, University of ‘Magna Græcia’ and Cancer Center, Catanzaro, Italy; 4Endocrine Unit, Massachusetts General Hospital, Boston, MA, USA and 5Callisto Pharmaceuticals Inc., New York, NY, USA Atiprimod (Atip) is a novel oral agent with anti-inflammatory tion.7,8 Atip inhibits the inflammatory response and preserves properties. Although its in vitro activity and effects on signaling bone integrity in murine models of rheumatoid arthritis (RA),9–12 in multiple myeloma (MM) have been previously reported, here targets macrophages, inhibits phospholipase A and C in rat we investigated its molecular and in vivo effects in MM. Gene 13,14 expression analysis of MM cells identified downregulation of alveolar macrophages and exhibits antiproliferative and 15–17 genes involved in adhesion, cell-signaling, cell cycle and bone antiangiogenic activities in human cancer models. Impor- morphogenetic protein (BMP) pathways and upregulation of tantly, we have previously reported that Atip inhibits MM cell genes implicated in apoptosis and bone development, follow- growth, induces caspase-mediated apoptosis, blocks the phos- ing Atip treatment. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Investigating the Mechanism of Hepatocellular Carcinoma Progression by Constructing Genetic and Epigenetic Networks Using NGS Data Identification and Big Database Mining Method
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 48 Research Paper Investigating the mechanism of hepatocellular carcinoma progression by constructing genetic and epigenetic networks using NGS data identification and big database mining method Cheng-Wei Li1, Ping-Yao Chang1, Bor-Sen Chen1 1Laboratory of Control and Systems Biology, National Tsing Hua University, Hsinchu, Taiwan Correspondence to: Bor-Sen Chen, email: [email protected] Keywords: DNA methylation, multiple potential drugs, hepatocarcinogenesis, miRNAs, principal network projection Received: February 19, 2016 Accepted: October 26, 2016 Published: November 04, 2016 ABSTRACT The mechanisms leading to the development and progression of hepatocellular carcinoma (HCC) are complicated and regulated genetically and epigenetically. The recent advancement in high-throughput sequencing has facilitated investigations into the role of genetic and epigenetic regulations in hepatocarcinogenesis. Therefore, we used systems biology and big database mining to construct genetic and epigenetic networks (GENs) using the information about mRNA, miRNA, and methylation profiles of HCC patients. Our approach involves analyzing gene regulatory networks (GRNs), protein-protein networks (PPINs), and epigenetic networks at different stages of hepatocarcinogenesis. The core GENs, influencing each stage of HCC, were extracted via principal network projection (PNP). The pathways during different stages of HCC were compared. We observed that extracellular signals were further transduced to -

Spotlight Review
Leukemia (2009) 23, 10–24 & 2009 Macmillan Publishers Limited All rights reserved 0887-6924/09 $32.00 www.nature.com/leu SPOTLIGHT REVIEW Bone marrow microenvironment and the identification of new targets for myeloma therapy K Podar, D Chauhan and KC Anderson Department of Medical Oncology, LeBow Institute for Myeloma Therapeutics, Dana Farber Cancer Institute, Jerome Lipper Multiple Myeloma Center, Harvard Medical School, Boston, MA, USA The development of multiple myeloma (MM) is a complex multi- Signaling cascades activated by cytokines, growth factors and/ step process involving both early and late genetic changes in or adhesion in MM cells include the Ras/Raf/MEK/MAPK- the tumor cell as well as selective supportive conditions by the k bone marrow (BM) microenvironment. Indeed, it is now well pathway, PI3K/Akt-pathway, the JAK/Stat3-pathway, the NF B- established that MM cell-induced disruption of the BM homeo- pathway and the Wnt-pathway. Promising intracellular targets stasis between the highly organized cellular and extracellular for novel therapies also include protein kinase C (PKC) and SPOTLIGHT compartments supports MM cell proliferation, survival, migra- heat-shock proteins (HSPs). Moreover, genomic profiling has tion and drug resistance through activation of various signaling now identified additional stage-specific intracellular targets, (for example, PI3K/Akt, JAK/Stat-, Raf/MEK/MAPK-, NFjB- and which are now under investigation as novel potential therapeutic Wnt-) pathways. Based on our enhanced understanding of the 2,4 functional importance of the MM BM microenvironment and its targets. inter-relation with the MM cell resulting in homing, seeding, Cell surface receptors include integrins, cadherins, selectins, proliferation and survival, new molecular targets have been syndecans, and the immunoglobulin superfamily of cell adhe- identified and derived treatment regimens in MM have already sion molecules including syndecan-1 (CD138), H-CAM (CD44), changed fundamentally during recent years. -

Alteration of Epileptogenesis Genes
Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics Alteration of Epileptogenesis Genes Amy R. Brooks-Kayal,*† Yogendra H. Raol,* and Shelley J. Russek‡ *Division of Neurology, Department of Pediatrics, University of Colorado Denver School of Medicine, †The Children’s Hospital, Aurora, Colorado 80045, and ‡Laboratory of Translational Epilepsy, Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, Boston, Massachusetts 02118 Summary: Retrospective studies suggest that precipitating expression and function have been reported in adult animals events such as prolonged seizures, stroke, or head trauma in- beginning immediately after prolonged seizures (status epilepticus crease the risk of developing epilepsy later in life. The process [SE]) and continue as animals become chronically epileptic. Pre- of epilepsy development, known as epileptogenesis, is associ- vention of GABAA receptor subunit changes after SE using viral ated with changes in the expression of a myriad of genes. One gene transfer inhibits development of epilepsy in an animal model, of the major challenges for the epilepsy research community suggesting that these changes directly contribute to epileptogen- has been to determine which of these changes contributes to esis. The mechanisms that regulate differential expression of epileptogenesis, which may be compensatory, and which may GABAA receptor subunits in hippocampus after SE have recently be noncontributory. Establishing this for any given gene -

Stat3 Inhibition Augments the Immunogenicity of B-Cell Lymphoma Cells, Leading to Effective Antitumor Immunity
Published OnlineFirst June 22, 2012; DOI: 10.1158/0008-5472.CAN-11-3619 Cancer Therapeutics, Targets, and Chemical Biology Research Stat3 Inhibition Augments the Immunogenicity of B-cell Lymphoma Cells, Leading to Effective Antitumor Immunity Fengdong Cheng1, Hongwei Wang1, Pedro Horna1,3, Zi Wang1, Bijal Shah1, Eva Sahakian1, Karrune V. Woan1, Alejandro Villagra1, Javier Pinilla-Ibarz1, Said Sebti2, Mitchell Smith4, Jianguo Tao1,3, and Eduardo M. Sotomayor1 Abstract Mantle cell lymphoma (MCL) is an aggressive and incurable subtype of B-cell non–Hodgkin lymphomas. Although patients often respond initially to first-line treatment with chemotherapy plus monoclonal antibodies, relapse and decreased response to further lines of treatment eventually occurs. Harnessing the immune system to elicit its exquisite specificity and long-lasting protection might provide sustained MCL immunity that could potentially eradicate residual malignant cells responsible for disease relapse. Here, we show that genetic or pharmacologic disruption of Stat3 in malignant B cells augments their immunogenicity leading to better þ activation of antigen-specific CD4 T cells and restoration of responsiveness of tolerized T cells. In addition, treatment of MCL-bearing mice with a specific Stat3 inhibitor resulted in decreased Stat3 phosphorylation in malignant B cells and anti-lymphoma immunity in vivo. Our findings therefore indicate that Stat3 inhibition may represent a therapeutic strategy to overcome tolerance to tumor antigens and elicit a strong immunity against MCL and other B-cell malignancies. Cancer Res; 72(17); 1–9. Ó2012 AACR. Introduction cell non–Hodgkin lymphomas (NHL; ref. 6). As such, novel – Previous studies in murine models of B-cell lymphoma non cross-resistant treatment modalities capable of improv- indicate that generation of effective anti-lymphoma immunity ing the response rate and more importantly able of sustaining requires (i) conversion of bone marrow–derived antigen-pre- these responses are greatly needed for patients with MCL. -
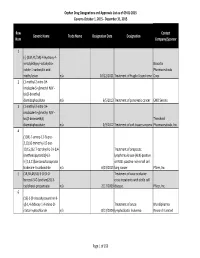
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

Milciclib and Sorafenib Synergistically Downregulate C-Myc to Suppress
Journal of Translational Science Research Article ISSN: 2059-268X Milciclib and sorafenib synergistically downregulate c-Myc to suppress tumor growth in an orthotopic murine model of human hepatocellular carcinoma Aastha Jindal1, Vaseem A Palejwala2, Marina Ciomei3, Anna Degrassi3, Gemma Texido3 and Kunwar Shailubhai1,2* 1Baruch S. Blumberg Institute, Pennsylvania Biotechnology Center, Doylestown, PA, USA 2Tiziana Lifesciences, 3805 Old Easton Road, Doylestown, PA, USA 3Nerviano Medical Sciences, 20014 Nerviano, Milan, Italy Abstract Complex heterogeneity in HCC pateints, due to multiple etiological factors, implies that monotherapy targeting a single mechanism might not be sufficient for sustained clinical response as another compensatory signaling could get activated to develop drug resistance. Thus, combination therapies utilizing drugs, preferentially with different mode of action, might be clinically more satisfactory. Milciclib (PHA-848125AC), a pan-inhibitor of cyclin dependent kinases (CDKs: CDK1, CDK2, CDK4, CDK5 and CDK7) and c-Src kinase, has shown anti-cancer activity in tumor models as well as in clinical studies with cancer patients, including HCC. Here, we present data demonstrating anti-HCC activity of milciclib in cell lines as well as in two different animal models HCC. Importantly, milciclib exhibited synergistic anti-HCC effect with sorafenib, a first-line tyrosine kinase inhibitor (TKI) drug for HCC. Oral treatment with milciclib inhibited HCC tumor growth in TG221 mice, a miR-221-induced transgenic HCC model. We also developed an orthotopic model, by implanting human HCC cells (MHCC97-H) in liver of nude mice, in which tumor growth can be monitored by measuring serum levels of human α-fetoprotein (hAFP) but not mouse AFP. -

Hallmarks of Gastrointestinal Neuroendocrine Tumours: Implications for Treatment
A Walenkamp et al. Hallmarks of GI-NETs: treatment 21:6 R445–R460 Review implications Hallmarks of gastrointestinal neuroendocrine tumours: implications for treatment Annemiek Walenkamp, Guillermo Crespo1, Felipe Fierro Maya2, Reidar Fossmark3, Peter Igaz4, Anja Rinke5, Gianluca Tamagno6, Giovanni Vitale7,8, Kjell O¨ berg9 and Tim Meyer10 Department of Medical Oncology, University Medical Centre Groningen, University of Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands 1Department of Medical Oncology, Hospital Universitario de Burgos, Avenida Islas Baleares 3, 09006 Burgos, Spain 2Department of Endocrine Oncology, National Cancer Institute, Bogota´ , Colombia 3Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, 7491 Trondheim, Norway 42nd Department of Medicine, Semmelweis University, 46, Szentkiralyi Street, H-1088 Budapest, Hungary 5Department of Gastroenterology, University Hospital Marburg, Baldinger Strasse, Marburg D-35043, Germany 6Department of General Internal Medicine, St Columcille’s Hospital, Loughlinstown - Co., Dublin, Ireland 7Department of Clinical Sciences and Community Health (DISCCO), University of Milan, Milan, Italy Correspondence 8Laboratory of Endocrine and Metabolic Research, Istituto Auxologico Italiano IRCCS, Via Zucchi 18, should be addressed Cusano Milanino (MI) 20095, Italy to A Walenkamp 9Department of Endocrine Oncology, University Hospital, Uppsala, Sweden Email 10UCL Cancer Institute, UCL, Huntley Street, London WC1E 6BT, UK [email protected] -

WO 2017/068349 Al 27 April 2017 (27.04.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/068349 Al 27 April 2017 (27.04.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 31/05 (2006.01) A61P 31/00 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/352 (2006.01) A61P 31/12 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 45/06 (2006.01) A61P 37/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, A61K 39/39 (2006.01) A61P 37/04 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, C07K 16/28 (2006.01) A61P 37/06 (2006.01) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, A61P 35/00 (2006.01) A61P 37/08 (2006.01) KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (21) International Application Number: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, PCT/GB20 16/053271 SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (22) International Filing Date: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 20 October 2016 (20.10.201 6) ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (30) Priority Data: TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, 15 18805.5 23 October 2015 (23. -

Atiprimod Blocks STAT3 Phosphorylation and Induces Apoptosis in Multiple Myeloma Cells
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central British Journal of Cancer (2005) 93, 70 – 80 & 2005 Cancer Research UK All rights reserved 0007 – 0920/05 $30.00 www.bjcancer.com Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells M Amit-Vazina1, S Shishodia1, D Harris1, Q Van1, M Wang2, D Weber2, R Alexanian2, M Talpaz1, BB Aggarwal1 *,1 and Z Estrov 1 2 Department of Bioimmunotherapy, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA; Department Lymphoma/Myeloma, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA Multiple myeloma (MM) accounts for 1 % of all cancer deaths. Although treated aggressively, almost all myelomas eventually recur Translational Therapeutics and become resistant to treatment. Atiprimod (2-(3-Diethylaminopropyl)-8,8-dipropyl-2-azaspiro[4,5] decane dimaleate) has exerted anti-inflammatory activities and inhibited oeteoclast-induced bone resorption in animal models and been well tolerated in patients with rheumatoid arthritis in phase I clinical trials. Therefore, we investigated its activity in MM cells and its mechanism of action. We found that Atiprimod inhibited proliferation of the myeloma cell lines U266-B1, OCI-MY5, MM-1, and MM-1R in a time- and dose-dependent manner. Atiprimod blocked U266-B1 myeloma cells in the G0/G1 phase, preventing cell cycle progression. Furthermore, Atiprimod inhibited signal transducer and activator of transcription (STAT) 3 activation, blocking the signalling pathway of interleukin-6, which contributes to myeloma cell proliferation and survival, and downregulated the antiapoptotic proteins Bcl-2, Bcl-X , and Mcl-1.